Background
Clinical intolerance to fructose was initially described in 1956. The following year, researchers reported a familial incidence of the disorder in several family members, postulating that the defect was a deficiency of hepatic fructose 1-aldolase. Within the next 4-5 years, the enzyme defect in aldolase B isozyme in the liver was demonstrated, and hereditary fructose intolerance (HFI) became recognized as a distinct clinical entity. The rapid early progress in the understanding of this disorder may have occurred because of the fairly dramatic and difficult-to-miss symptoms associated with fructose ingestion. These symptoms include vomiting, hypoglycemia, failure to thrive, cachexia, hepatomegaly, jaundice, coagulopathy, coma, renal Fanconi syndrome, and severe metabolic acidosis (in part due to lactic acidosis). See the image below.
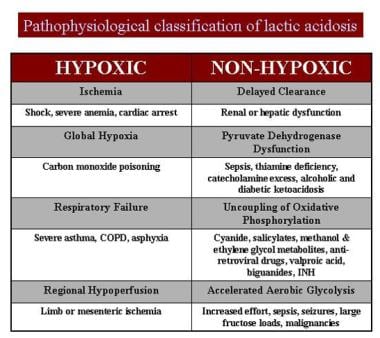 Pathophysiologic classification of lactic acidosis.
Pathophysiologic classification of lactic acidosis.
Pathophysiology
Affected individuals are completely asymptomatic until they ingest fructose. Thus, homozygous neonates remain clinically well until confronted with dietary sources of fructose. Although lactose is the carbohydrate base in most infant formulas, some (eg, soy formulas) contain sucrose, a fructose-glucose disaccharide that may cause symptoms. [1] The biochemistry of hereditary fructose intolerance is complex for 2 reasons: (1) 3 isozymes of aldolase (A, B, C) exist, of which aldolase B is expressed exclusively in the liver, kidney, and intestine, and (2) aldolase B mediates 3 separate reactions (ie, cleavage of fructose 1-phosphate [F-1-P]; cleavage of fructose 1,6-diphosphate; and condensation of the triose phosphates, glyceraldehyde phosphate, and dihydroxyacetone phosphate to form fructose 1,6-diphosphate). [2, 3, 4]
In normal cellular conditions, the primary enzymatic activity of aldolase B is to cleave fructose diphosphate (FDP), which forms rather than condenses the triose phosphate compounds. Here, the enzyme is central to the glycolytic pathway. Because the reaction is reversible, aldolase B is an essential enzyme in the process of gluconeogenesis (which is, in some respects, a reversal of glycolysis). The absence of the latter function readily explains the clinical hypoglycemia in individuals with hereditary fructose intolerance.
Reduced cleavage of F-1-P leads to its cellular accumulation and fructokinase inhibition, causing free fructose accumulation in the blood. A generally accepted consequence of this sequence is a dramatic change in the ATP-adenosine monophosphate (AMP) cellular ratio, with a resultant acceleration in production of uric acid. This accounts for the hyperuricemia observed during an acute episode. Competition between urate and lactate for renal tubule excretion accounts for the lactic acidemia.
The cause of severe hepatic dysfunction remains unknown but may be a manifestation of focal cytoplasmic degeneration and cellular fructose toxicity. [5] The development of a mouse knock-out model for HFI may provide additional information in this regard. [6] The cause of renal tubular dysfunction also remains unclear; patients with renal tubular dysfunction primarily present with a proximal tubular acidosis complicated by aminoaciduria, glucosuria, and phosphaturia. Thus, in an infant who is homozygous for fructose 1-aldolase deficiency, fructose ingestion triggers a cascade of biochemical events that result in severe clinical disease.
Epidemiology
Frequency
United States
Although the true prevalence has not been established, hereditary fructose intolerance may be more common than originally believed; many asymptomatic affected people may simply avoid the ingestion of most or all sweets. The prevalence has been estimated to be as high as 1 case per 20,000 individuals.
International
The prevalence of hereditary fructose intolerance in central Europe has been reported to be 1 case per 26,100 individuals. [7, 8]
Mortality/Morbidity
Morbidity is implicit in untreated patients. Hypoglycemia and acidosis may act together to cause organ shock or coma. Ongoing hepatocellular insult may result in cirrhosis and eventual hepatic failure. Failure to thrive progressing to cachexia is the rule. Mortality may result from any or all of the above conditions.
Sex
Hereditary fructose intolerance is an autosomal recessive trait that is equally distributed between the sexes.
Age
In many infants, the age at symptom onset leads to the diagnosis. An accurate dietary history can indicate a link between the introduction of fruits into the diet and symptom onset. [9] Adult-onset disease has been reported, [10] although whether such patients avoided fructose ingestion, thus avoiding clinical symptoms, or the latter truly did not manifest until adulthood, is arguable.
Prognosis
The prognosis is excellent for infants who receive rapid diagnosis and treatment.
In the absence of substantial hepatic damage, life expectancy is normal.
Patient Education
Parents must receive genetic counseling as part of their education in the care of the child.
Stress the importance of input from a nutritionist and the essential nature of a cooperative relationship in the long-term care of the child.
-
Pathophysiologic classification of lactic acidosis.

