Background
Hereditary elliptocytosis and hereditary pyropoikilocytosis are congenital hemolytic disorders in which erythrocytes either are elongated into an oval form or are irregularly shaped (see images below). [1] Transmission is primarily via an autosomal dominant pattern, but de novo mutations, recessive inheritance, and X-linkage have also been described, [2] with next-generation sequencing (NGS) permitting a wider variety of mutations to be identified. [3]
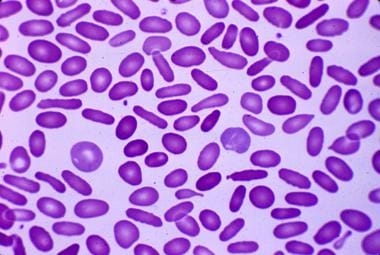 Cigar-shaped erythrocytes seen in hereditary elliptocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
Cigar-shaped erythrocytes seen in hereditary elliptocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
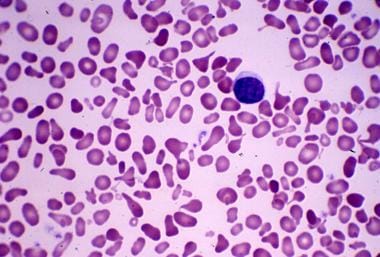 Bizarre RBC morphology seen in hereditary pyropoikilocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
Bizarre RBC morphology seen in hereditary pyropoikilocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
These disorders are characterized by clinical, biochemical, and genetic heterogeneity. Numerous molecular defects have been implicated in the pathogenesis of these disorders. Clinical manifestations range from an asymptomatic carrier state to severe hemolytic anemia. Expressivity is variable, with members of the same family with the same genetic defect exhibiting different clinical courses. Additionally, an individual's frequency and severity of hemolysis may change with time.
Pathophysiology
Hereditary elliptocytosis and its related disorders are caused by mutations that disrupt the RBC cytoskeleton, a multiprotein complex responsible for the elasticity and durability of the circulating erythrocytes. RBCs must be adequately resilient and sufficiently flexible to withstand shearing forces as they pass through the microcirculation.
The normal RBC membrane consists of a lipid bilayer, which contains proteins and glycans.
Spectrin tetramers form a large part of the cytoskeletal framework and are composed of heterodimers of alpha and beta subunits. These are tethered to the plasma membrane proteins AE1 (band 3) and glycophorin C through the ankyrin/protein 4.2 complex and through protein 4.1R and its associated actin filaments. Defects in horizontal interactions result in hereditary elliptocytosis.
The image seen below depicts the complexity of the RBC membrane.
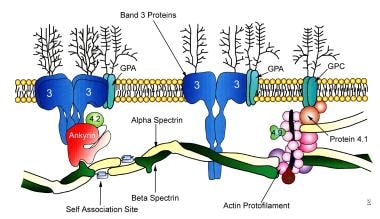 Schematic diagram of the components of the RBC membrane. Hereditary elliptocytosis can result from defects of alpha or beta spectrin or from a defective spectrin-actin-protein 4.1R junctional complex.
Schematic diagram of the components of the RBC membrane. Hereditary elliptocytosis can result from defects of alpha or beta spectrin or from a defective spectrin-actin-protein 4.1R junctional complex.
A crystal structure of the spectrin tetramerization domain complex has been determined, and information gleaned from studying it is expected to further elucidate the molecular basis of hereditary hemolytic anemias.
In hereditary elliptocytosis, circulating erythrocytes undergo a progressive transformation from a normal discocyte to an elliptocyte. Most cases of hereditary elliptocytosis are the result of mutations of alpha spectrin or beta spectrin. A minority of cases are due mutations of protein 4.1R, ankyrin, or glycophorin C. The result of these mutations is a mechanically unstable membrane that is less tolerant of shear stress and susceptible to permanent deformation.
RBC precursors in common hereditary elliptocytosis are round but become more elliptical as they age. Normal RBCs also undergo repeated elliptical deformation during circulation. Normal erythrocytes, however, regain their discoid shape. Erythrocytes in hereditary elliptocytosis lack elasticity and remain elliptical. Elliptocytes and poikilocytes are postulated to be permanently fixed in their abnormal shape because the weakened membrane interactions facilitate skeletal reorganization after prolonged or repetitive cellular deformation. This results in mechanical instability and susceptibility to fragmentation and lysis. Splenic sequestration of these abnormal RBCs is the dominant cause of decreased erythrocyte survival in hereditary elliptocytosis.
Some of the more severe forms of hereditary elliptocytosis are associated with marked poikilocytosis or variation in RBC shape. This is deemed hereditary pyropoikilocytosis. Hereditary pyropoikilocytosis is characterized by bizarre RBC morphology similar to that seen in thermal burns. Blood smear findings are most notable for fragmented erythrocytes and microspherocytes. Elliptocytes are present but in fewer numbers. The RBCs demonstrate features of decreased deformability and increased membrane fragmentation.
Hereditary pyropoikilocytosis and more severe cases of hereditary elliptocytosis likely result from coinheritance of a typical hereditary elliptocytosis mutation and a relatively common but clinically silent alpha-spectrin gene allele alpha-LELY. This is homozygous or compound heterozygous inheritance. [2]
An acquired form of elliptocytosis has been identified in several bone marrow disorders, including fibrosis, myelophthisis, and myelodysplasia. [4]
Southeast Asian ovalocytosis is form of elliptocytosis. This condition is unique because a single mutation of band 3 (anion exchanger 1) is responsible for the defect. All individuals with Southeast Asian ovalocytosis are heterozygotes for band 3 gene mutations because the homozygous form is lethal in utero.
Epidemiology
Frequency
United States
The true number of cases is unknown because the clinical severity of this group of disorders is heterogeneous and many patients are asymptomatic and without anemia. There is a strong association between hereditary elliptocytosis and hereditary pyropoikilocytosis in families. Patients with hereditary pyropoikilocytosis may have relatives with undiagnosed hereditary elliptocytosis.
Clinically apparent elliptocytic disorders are present in approximately 1 in 2000-4000 individuals.
International
Worldwide, the incidence varies widely. The conditions are seen in increased frequency in Southeast Asia, Africa, and the Mediterranean. It is most common in malaria-endemic regions. The prevalence of hereditary elliptocytosis in West Africa, for example, approaches 2%. [5] The prevalence of Southeast Asian ovalocytosis may be as high as 25% in several Southeast Asian ethnic groups. These groups are primarily found in the Philippines, southern Thailand, Malaysia, Papua New Guinea, Indonesia, Borneo, Brunei, Cambodia, and in native Australians and certain native South Africans. [6]
Mortality/Morbidity
Morbidity in these disorders depends on the frequency and degree of hemolytic anemia. The clinical phenotype ranges from asymptomatic carrier status to severe transfusion-dependent, and even fatal, hemolytic anemia or hydrops fetalis. Individuals with chronic hemolysis may have complications such as jaundice, splenomegaly, aplastic crises, and early gallbladder disease. Southeast Asian ovalocytosis is homozygous lethal to fetuses. Otherwise, mortality is rare.
Race
Hereditary elliptocytosis and hereditary pyropoikilocytosis are more common in individuals of West African, Central African, Mediterranean, and Southeast Asian descent. This distribution parallels the geographic distribution of malaria.
Sex
No sex predilection is observed.
Age
This condition may present at any age with hemolytic anemia and the characteristic morphological abnormalities. In anemic pregnant women, it is important to diagnose hereditary elliptocytosis because neonates of affected women are at risk of clinically significant perinatal hemolysis with severe anemia and hyperbilirubinemia. [7] Hydrops fetalis may also occur.
-
Cigar-shaped erythrocytes seen in hereditary elliptocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.
-
Schematic diagram of the components of the RBC membrane. Hereditary elliptocytosis can result from defects of alpha or beta spectrin or from a defective spectrin-actin-protein 4.1R junctional complex.
-
Bizarre RBC morphology seen in hereditary pyropoikilocytosis. Courtesy of Jean A. Shafer, BS, MA, Assistant Professor of Hematology and Pathology at the University of Rochester School of Medicine and Dentistry.




