Practice Essentials
Gaucher disease is a rare genetic disorder characterized by the deposition of glucocerebroside in cells of the macrophage-monocyte system. The disorder results from the deficiency of the enzyme glucocerebrosidase. [1]
Signs and symptoms
While Gaucher disease manifests with vast clinical heterogeneity, it traditionally has been differentiated into the following 3 clinical subtypes, delineated by the absence or presence of neurologic involvement and its progression [1] :
-
Type 1 - Nonneuronopathic Gaucher disease
-
Type 2 - Acute neuronopathic Gaucher disease
-
Type 3 - Chronic neuronopathic Gaucher disease
Patients with type 1 disease commonly present with painless splenomegaly, anemia, or thrombocytopenia. They may have chronic fatigue, hepatomegaly (with or without abnormal liver function test findings), bone pain, or pathologic fractures and may bruise easily because of thrombocytopenia. Bleeding secondary to thrombocytopenia may manifest as nosebleeds, bruising, or both.
Patients with type 2 disease may present prenatally, at birth or during infancy with increased tone, seizures, strabismus, and organomegaly. Failure to thrive, swallowing abnormalities, oculomotor apraxia, hepatosplenomegaly, and stridor due to laryngospasm are typical in infants with type 2 disease.
Patients with type 3 disease, in addition to organomegaly and bony involvement, present with neurologic involvement, most often including slowing of the horizontal saccadic eye movements. The neurologic manifestations may be mild or present subtly in infancy to early childhood.
See Clinical Presentation for more detail.
Diagnosis
Diagnosis can be confirmed through measurement of glucocerebrosidase activity in peripheral blood leukocytes. A finding of less than 15% of mean normal activity is diagnostic. Minor elevations of liver and angiotensin-converting enzyme levels are common. DNA analysis is also commonly used to establish the diagnosis based on presence of 2 mutant alleles, especially in diagnostic panels. [1]
See Workup for more detail.
Management
Enzyme replacement therapy (ERT) is indicated for patients with type 1 and type 3 Gaucher disease who exhibit clinical signs and symptoms of the disease, including anemia, thrombocytopenia, skeletal disease, or visceromegaly. Substrate reduction therapy (SRT) is an alternative treatment for appropriate adult patients with type 1 Gaucher disease. SRT works via global reduction of glucosylceramide synthase and has been shown to yield outcomes similar to those of ERT in adults. ERT sometimes is started in patients with type 2 GD, as often there can be a question regarding disease type and progression, and to delay may have significant impact on patient outcomes. [1]
See Treatment and Medication for more detail.
Background
Gaucher disease is a lipid storage disease characterized by the deposition of glucocerebroside in cells of the macrophage-monocyte system. The disorder results from the deficiency of a specific lysosomal hydrolase, glucocerebrosidase (also termed acid beta-glucosidase, glucosylceramidase). The disease is best characterized as a continuum of phenotypes. The severity widely varies; some patients present in childhood with virtually all the complications of Gaucher disease, whereas others remain asymptomatic into the eighth decade of life. [1]
Gaucher disease has traditionally been divided into the following 3 clinical subtypes, delineated by the absence or presence of neurologic involvement and its progression [1] :
-
Type 1 - Nonneuronopathic form
-
Type 2 - Acute neuronopathic form
-
Type 3 - Chronic neuronopathic form
However, some cases do not fit precisely into one of these categories, and the disease should be viewed as a spectrum of symptoms.
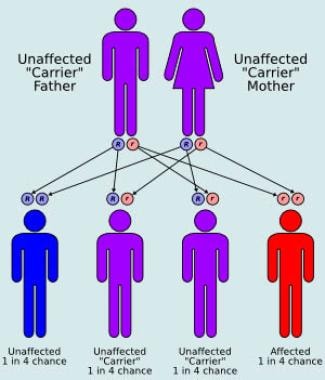 Autosomal recessive inheritance pattern.
Autosomal recessive inheritance pattern.
Type 1 Gaucher disease is more common among individuals with Ashkenazi Jewish heritage, although all types are panethnic in their distribution.
Pathophysiology
Glucosylceramide, the accumulated glycolipid, is primarily derived from the phagocytosis and degradation of senescent leukocytes and erythrocyte membranes. The glycolipid storage gives rise to the characteristic Gaucher cells, macrophages engorged with lipid with a crumpled–tissue-paper appearance and displaced nuclei. The factors that contribute to neurologic involvement in patients with types 2 and 3 disease are still unknown but may be related to the accumulation of a cytotoxic glycolipid, glucosylsphingosine, in the brain due to the severe deficiency of glucocerebrosidase activity or to neuroinflammation. [1, 2]
Glucosylceramide accumulation in the bone marrow, liver, spleen, lungs, and other organs contributes to pancytopenia, massive hepatosplenomegaly, and, at times, diffuse infiltrative pulmonary disease. Progressive infiltration of Gaucher cells in the bone marrow may lead to thinning of the cortex, pathologic fractures, bone pain, bony infarcts, and osteopenia. These bony features may also be related to cytokines produced by macrophages.
Dramatic changes to the ceramide-to-glucosylceramide ratio can affect the barrier formation in the epidermal layer of the skin, leading to ichthyosis or a collodion skin presentation, seen at times in babies who are severely affected (those with type 2).
Epidemiology
Frequency
United States
Type 1 Gaucher disease is more common among Jewish people of Eastern European origin; the carrier frequency in these individuals is approximately 1 in 15 population, whereas the disease frequency is 1 in 855 population. Gaucher disease is rare in the non-Jewish population, with an estimated frequency of 1 per 40,000 population.
International
Internationally, the disease frequency is similar to that in the United States, except for areas of the world with large Ashkenazi Jewish populations. Most patients worldwide are non-Jewish. As many as 60% of patients of Ashkenazi origin are estimated to be homozygous for the mild N370S mutation, which accounts for 75% of disease alleles in this population. Many individuals with this genotype never seek medical attention, contributing to an underestimation of the disease frequency. Type 3 disease is more common in the Norrbottnian region of Sweden (1 per 50,000 population), which has been traced to a common founder in the 17th century. Type 3 Gaucher disease also appears to be the most frequent form in areas of Asia.
Mortality/Morbidity
Mortality and morbidity varies with the different types. [3]
Type 1 Gaucher disease may present in childhood with hepatosplenomegaly, pancytopenia, and skeletal disease, although there is striking clinical variability in disease severity. Prior to the advent of treatment, bleeding and hepatic complications were more common, and patients with severe splenomegaly would routinely undergo splenectomy and have severe, sometimes fatal, bleeding complications. This risk has been dramatically reduced with the development of enzyme replacement therapy (ERT). Bone disease was a common cause of morbidity but is seen less frequently in treated patients. [1, 4, 5]
Type 2 Gaucher disease causes rapidly progressive neurovisceral storage disease and death during infancy or during the first years of life. A subset of this type, associated with congenital ichthyosis and hydrops fetalis, is described as neonatal lethal and results in perinatal or in utero death. [1, 4, 5]
Type 3 Gaucher disease often is a less rapidly progressive neurovisceral storage disease, but it does have the largest phenotypic variation of among the GD spectrum. Various associated clinical courses have been reported, some of which cause death in childhood or early adulthood. Others, when treated, have a clinical progression like that of type 1 Gaucher disease and have very subtle neurologic findings. [1, 4, 5]
Race
All forms of Gaucher disease are panethnic. Type 1 Gaucher disease is one of the most common lysosomal storage diseases and is the most prevalent genetic disorder in individuals of Ashkenazi Jewish descent. Type 3 disease is more common in the Norrbottnian region of Sweden and parts of Asia.
Sex
All 3 types of Gaucher disease are autosomal recessively inherited and have an equal sex distribution.
Age
Patients with type 1 Gaucher disease may present in childhood with hepatosplenomegaly, pancytopenia, and crippling skeletal disease. Some patients are not diagnosed until adulthood, when they present with low blood counts, bleeding events, incidental splenomegaly, or bone involvement, whereas others are diagnosed in the seventh to ninth decades of life after an incidental finding of thrombocytopenia or splenomegaly. Many affected individuals never develop signs or symptoms and do not seek medical attention. Types 2 and 3 Gaucher disease typically present in early childhood. With an increased awareness of the connection between GD and Parkinson Disease, some patients have been diagnosed in Movement Disorder clinics at a later age.
Prognosis
Some individuals with type 1 Gaucher disease have few manifestations and a normal life expectancy without any intervention. The prognosis of symptomatic type 1 or type 3 Gaucher disease in patients who receive treatment is very good, with a decrease in organomegaly and an eventual rise in hemoglobin levels and platelet counts.
One study estimated life expectancy from birth in individuals with type 1 Gaucher disease to be 68 years, compared with 77 years in the reference population, [6] although this study was based on one industry-supported patient registry that may not have included many patients with milder cases who do not require therapy.
Skeletal disease is slow to respond to ERT, is generally more pronounced if a patient has undergone a splenectomy, and varies widely. Early treatment may decrease the frequency of bony involvement.
Some patients describe symptomatic improvement within the first year of treatment, although a much longer period of ERT is required to achieve a radiologic response.
The prognosis of type 2 Gaucher disease is universally poor, and depending on the age at presentation and the aggressiveness of management, life expectancy varies from several months to several years. [7]
The prognosis of type 3 Gaucher disease depends on the severity of disease and age of onset of therapy. Most patients with progressive myoclonic epilepsy associated with GD3 do not fare well. Also those with the cardiac variant associated specifically with genotype D409H/D409H, GD3C often develop valvular calcifications and die of cardiac complications in the second or third decade of life.
Patient Education
Patients with Gaucher disease and their families require education regarding the disease manifestations, variability in symptoms and disease progression, and potential complications. In addition, they should be counseled regarding recurrence risks.
There are several ethical considerations facing patients with Gaucher disease and their medical team. With the rise of newborn screening covering lysosomal storage diseases, an increasing number of Gaucher patients are identified when asymptomatic. The question of when to begin treatment is debated and is particularly relevant with the pediatric patients who are asymptomatic. The decision whether to treat patients with type 2 GD is controversial. As there is often a diagnostic challenge discerning between acute and chronic neuronopathic Gaucher disease, current recommendation is to begin ERT if there is any question of diagnosis.
-
Autosomal recessive inheritance pattern.






