Background
As with all hexose sugars, metabolism of ingested galactose requires an initial phosphorylation of the molecule using adenosine triphosphate (ATP). Unlike the metabolism of glucose, which ordinarily depends on the activity of hexokinase with a wide substrate-specificity to carry out this reaction, substrate-specific galactokinase activity exclusively phosphorylates galactose. [1]
In 1965, galactokinase deficiency was first identified in a patient who presented with cataracts and galactosuria that developed upon drinking milk. The concurrence of cataracts and galactosuria in a single individual suggested the possibility of a new type of galactosemia. This presentation differed from that of classic galactosemia in many important aspects; neither hepatosplenomegaly nor signs of mental retardation were present. When the researchers realized that the patient did not accumulate galactose-1-phosphate despite the accumulated galactose, the patient's underlying defect was deduced as the lack of the enzyme mediating 1-phosphorylation of galactose.
Pathophysiology
See the image below.
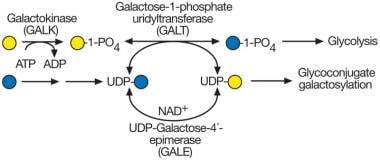 UDP-galactose synthesis and galactosemia. The most common form of galactosemia is due to a deficiency of galactose-1-phosphate uridyltransferase (GALT). This enzyme normally uses galactose-1-phosphate derived from dietary galactose. In the absence of GALT, galactose-1-phosphate accumulates, along with excessive galactose and its oxidative and reductive products galactitol and galactonate (not shown). UDP-galactose synthesis may also be impaired in the absence of GALT but not completely because UDP-galactose-4′-epimerase (GALE) can form UDP-galactose from UDP-glucose and can supply the donor to galactosyltransferases required for normal glycoconjugate biosynthesis.
UDP-galactose synthesis and galactosemia. The most common form of galactosemia is due to a deficiency of galactose-1-phosphate uridyltransferase (GALT). This enzyme normally uses galactose-1-phosphate derived from dietary galactose. In the absence of GALT, galactose-1-phosphate accumulates, along with excessive galactose and its oxidative and reductive products galactitol and galactonate (not shown). UDP-galactose synthesis may also be impaired in the absence of GALT but not completely because UDP-galactose-4′-epimerase (GALE) can form UDP-galactose from UDP-glucose and can supply the donor to galactosyltransferases required for normal glycoconjugate biosynthesis.
An appreciation of the differences between the enzyme deficiencies and their clinical manifestations is key to understanding the pathophysiology of galactokinase and galactose-1-phosphate uridyltransferase galactosemias. [2] Whereas vomiting, failure to thrive, jaundice, hepatomegaly, and cataracts are characteristic of the onset of transferase-deficient galactosemia, cataract development is usually the only symptom observed in an infant with kinase deficiency. In people with transferase-deficient galactosemia, galactose-1-phosphate accumulates; in those with kinase deficiency, galactose-1-phosphate cannot be produced. Galactose-1-phosphate is assumed to be the substance that causes the devastating manifestations seen in people with classic galactosemia. Note that this assumption lacks definitive proof despite the intrinsic and compelling logic.
In contrast, the mechanism that produces galactose-related cataracts is understood fairly well. The lens of the eye contains the aldose reductase enzyme. When presented with accumulated galactose, this enzyme reduces the aldehydic end group and produces galactitol, the analogous sugar alcohol. This compound exerts osmotic pressure within the lens because it slowly diffuses. While the induced lenticular swelling is not solely responsible for subsequent cataract formation, most researchers believe that the inciting event is galactitol rather than galactose-1-phosphate accumulation. The evidence favors this view because patients with galactokinase deficiency who cannot produce galactose-1-phosphate still form cataracts. [3]
While patients who are deficient in galactokinase accumulate galactitol in the liver at rates comparable to those with transferase-deficient galactosemia, only the latter display evidence of hepatic damage. Hence, much remains to be learned about the pathophysiologic implications of galactose metabolic impairment.
Epidemiology
Frequency
United States
Traditionally, most newborn screening programs were designed to identify transferase deficiency; consequently, accumulated galactose in submitted blood samples could be missed. [4] However, with the advent of cost-effective tandem mass spectrometry (MS/MS) newborn screening technology, which has been widely adopted in the United States and the rest of the developed world, screening for galactokinase deficiency is improving.
At present, 17 states in the United States either specifically include MS/MS in their newborn screening, or they use screening technology that is likely to detect galactokinase deficiency. [5, 6] Accordingly, because such screening technology is relatively recent, the data are insufficient to provide an accurate assessment of the prevalence of galactokinase deficiency; however, the estimated range is 1 per 50,000-100,000 live births.
International
The prevalence among certain Eastern European populations, in particular the Romani (Gypsy) population, is estimated to be approximately 1 per 10,000. The Romani people generally possess a mutation known as P28T, considered the founder mutation.
Mortality/Morbidity
The literature indicates no risk of mortality. Morbidity is limited to cataract formation in untreated individuals, although rare cases of pseudotumor cerebri have been reported. Both resolve with effective therapy. Mental retardation and hepatic damage are not associated with galactokinase deficiency. [7]
Sex
As an autosomal recessive condition, the disorder is distributed equally between sexes.
Age
Because galactokinase deficiency is a genetic disease, it is present from conception and may be discovered at birth through the presence of congenital cataracts.
-
UDP-galactose synthesis and galactosemia. The most common form of galactosemia is due to a deficiency of galactose-1-phosphate uridyltransferase (GALT). This enzyme normally uses galactose-1-phosphate derived from dietary galactose. In the absence of GALT, galactose-1-phosphate accumulates, along with excessive galactose and its oxidative and reductive products galactitol and galactonate (not shown). UDP-galactose synthesis may also be impaired in the absence of GALT but not completely because UDP-galactose-4′-epimerase (GALE) can form UDP-galactose from UDP-glucose and can supply the donor to galactosyltransferases required for normal glycoconjugate biosynthesis.

