Practice Essentials
Hemophagocytic lymphohistiocytosis (HLH) is a rare but potentially fatal disease of normal but overactive histiocytes and lymphocytes that commonly appears in infancy, although it has been seen in all age groups. Fever, hepatosplenomegaly, pancytopenia, lymphadenopathy, and rash often comprise the initial presentation. Rapid diagnosis and early treatment are crucial.
Signs and symptoms
Clinical findings include the following:
-
Fever
-
Hepatosplenomegaly
-
Lymphadenopathy
-
Jaundice
-
Rash (eg, erythroderma, generalized purpuric macules and papules, and morbilliform eruptions)
-
Easy bruisability and pallor
-
Swollen or hemorrhagic gums that can result in tooth loss
-
Central nervous system involvement, such as seizures, ataxia, hemiplegia, mental status changes, and irritability
-
Abdominal pain, vomiting, and diarrhea
-
Anorexia with or without weight loss
-
Feeding problems (especially prominent in infants)
-
Failure to thrive
See Presentation for more detail.
Diagnosis
The following 5 criteria set forth by the Histiocyte Society must be met to establish a diagnosis of HLH [1] :
-
Fever
-
Splenomegaly
-
Pancytopenia
-
Hypofibrinogenemia or hypertriglyceridemia
-
Hemophagocytosis
Because natural killer cell function or activity is decreased in up to 90% of patients with HLH, it is one of the most useful laboratory tests.
No specific imaging patterns are diagnostic.
A skin biopsy can assist in ruling out other systemic and potentially neoplastic diseases; however, the findings are usually not diagnostic and only rarely show hemophagocytosis. Because hemophagocytosis must be demonstrated in the bone marrow, spleen, or lymph nodes, appropriate specimens should be collected for documentation. See the image below.
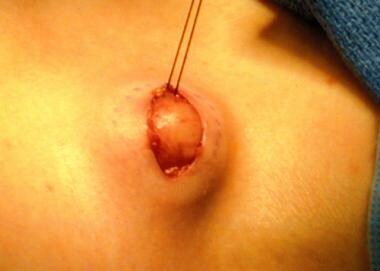 A lymph node biopsy is performed. Note that a marking pen has been used to outline the node before removal and that a silk suture has been used to provide traction to assist the removal.
A lymph node biopsy is performed. Note that a marking pen has been used to outline the node before removal and that a silk suture has been used to provide traction to assist the removal.
See Presentation and Workup for more detail.
Management
Initial therapy in patients with HLH consists of etoposide and dexamethasone for 8 weeks. Cyclosporine may be added to the initial regimen.
Resolved nonfamilial HLH does not require continuation of the regimen unless disease reactivation occurs after completion of the initial therapy or unless patients are undergoing bone marrow transplantation (BMT). For children with persistent nonfamilial disease or familial disease, continuation therapy with intravenous etoposide infusions, dexamethasone pulses, and oral cyclosporine is instituted at week 9 from the start of initial treatment.
Emapalumab is indicated for primary HLH in adults and children with refractory, recurrent, or progressive disease or who are intolerant to conventional HLH therapy. [2]
BMT is performed when a suitable donor can be found and the patient’s condition is stable.
See Treatment and Medication for more detail.
Background
Cutaneous involvement occurs in as many as 65% of patients with hemophagocytic lymphohistiocytosis (HLH). [3] Varied skin manifestations of HLH are noted, including erythroderma, generalized purpuric macules and papules, and morbilliform eruptions. Detection of cutaneous involvement can assist in the initial diagnosis of HLH and can potentially signify recurrences. Hemophagocytic lymphohistiocytosis may be viewed as a marker for underlying cancer, which in adults is most often a lymphoma that may be rapidly progressive. [4]
Primary hemophagocytic lymphohistiocytosis (ie, familial erythrophagocytic lymphohistiocytosis [FEL]), an inherited form of hemophagocytic lymphohistiocytosis syndrome, is a heterogeneous autosomal recessive disorder found to be more prevalent with parental consanguinity. Secondary hemophagocytic lymphohistiocytosis (ie, acquired hemophagocytic lymphohistiocytosis) occurs after strong immunologic activation, such as that which can occur with systemic infection, immunodeficiency, or underlying malignancy. Both forms are characterized by the overwhelming activation of normal T lymphocytes and macrophages, invariably leading to clinical and hematologic alterations and death in the absence of treatment. [5]
Drug reaction with eosinophilia and systemic symptoms (DRESS) is also a hypersensitivity reaction with overlapping syndromes with HLH, specifically dermatitis, lymphadenopathy, fever, eosinophilia, and visceral organ involvement. [6, 7] DRESS is associated with reactivation of herpes viruses with activated CD8+ T lymphocytes directed against them. Thus, patients with DRESS should be evaluated for the development of HLH, including for reactivation of human herpes viruses such HHV-6, HHV-7 and Epstein-Barr virus (EBV), and coagulation function evaluations. [8]
The history of this disorder, its molecular basis, and treatment options are noteworthy. [9] Almost 60 years has passed since Scottish pediatricians James Farquhar and Albert Claireaux, both of the University of Edinburgh, noticed the familial recurrence of this disorder affecting male and female siblings aged 2 months, causing fever, cytopenia, hepatosplenomegaly, and rapidly death despite treatment with antibiotics and steroids. [10]
Pathophysiology
The pathologic hallmark of this disease is the aggressive proliferation of activated macrophages and histiocytes, which phagocytose other cells, namely red blood cells (RBCs), white blood cells (WBCs), and platelets, leading to the clinical symptoms. The uncontrolled growth is nonmalignant and does not appear clonal in contrast to the lineage of cells in Langerhans cells histiocytosis (histiocytosis X). The spleen, lymph nodes, bone marrow, liver, skin, and membranes that surround the brain and spinal cord are preferential sites of involvement. [11] This disorder may be viewed as a highly stimulated, but ineffective, immune response to antigens, which results in life-threatening cytokine storm and inflammatory reaction. [12]
Over the past 2 decades, the underlying pathophysiology of hemophagocytic lymphohistiocytosis has been characterized, although the processes are not entirely understood. A current accepted theory involves an inappropriate immune reaction caused by proliferating and activated T cells associated with macrophage activation and inadequate apoptosis of immunogenic cells. [13] Although the precise mechanism remains unclear, many research teams propose convincing pictures for the role of perforin and natural killer (NK) cells in the hemophagocytic lymphohistiocytosis subtypes. [14, 15, 16]
Perforin or pore-forming protein (PFP), gene map location 10q22, is one of the major cytolytic proteins of granules contained in cytotoxic cells. [17] When activated by a challenge, NK cells release granules that contain perforin and granzymes, which form pores in the target cell membrane and cause osmotic lysis and protein degradation, respectively. Additionally, the endocytotic and exocytotic mechanisms may also be affected. [18] Patients with perforin deficiency may have impaired defenses against intracellular pathogens and cancers, as has been demonstrated in animal models.
Although the mechanism is yet to be determined, decreased NK cell activity results in increased T-cell activation and expansion, with resulting production of large quantities of cytokines, including interferon gamma (IFNg), tumor necrosis factor-α (TNF-α), and granulocyte-macrophage colony-stimulating factor (GM-CSF). This causes sustained macrophage activation and tissue infiltration as well as production of interleukin-1 (IL-1) and interleukin-6 (IL-6). The resulting inflammatory reaction causes extensive damage and the associated symptoms. [19]
While mutations in STX11 are said to be responsible for much of the familial hemophagocytic lymphohistiocytosis in the Middle East, one survey found such mutations in only 1% of North American patients. [20]
The familial form is a rare autosomal recessive disorder that has been classified into 6 different types based on genetic linkage analysis and chromosomal localization; 5 specific genetic defects have been identified, which account for approximately 90% of all patients. [21] Type 1 is due to a gene defect on chromosome 9, type 2 is due to mutations in the perforin gene, type 3 is due to mutations in the Munc-13-4 (UNC13D) gene, type 4 is due to mutations in the syntaxin 11 (STX11) gene, and type 5 is due to mutations in the gene encoding syntaxin binding protein 2 (STXBP-2).
Epstein-Barr virus (EBV) is the pathogen that most commonly triggers infection-associated hemophagocytic lymphohistiocytosis. One study showed a vulnerability to EBV infection in Korean children. [22] Epstein-Barr virus also infects CD8(+) T cells in EBV-associated hemophagocytic lymphohistiocytosis. [23, 24] Demonstrating these cells by their immunophenotypic characteristics may aid in the diagnosis of EBV-associated hemophagocytic lymphohistiocytosis. However, in a Brazilian sampling of 50 patients, 34 adults and 16 children, EBV infection was found to be less common in adults than in children. [25]
Hemophagocytic lymphohistiocytosis may be a complication of dengue, although this association is unusual. [26, 27] Scrub typhus may also be linked. [28]
The tick-borne parasitic infection babesiosis can have findings ranging from mild anemia to hemophagocytic lymphohistiocytosis. [29] One should also consider ehrlichiosis as a possible trigger of hemophagocytic lymphohistiocytosis. [30]
Hemophagocytic lymphohistiocytosis may be a complication of the DRESS syndrome. [8]
Epidemiology
International statistics
Incidence is reported to be 1.2 cases per million persons per year. However, unpublished observations estimate that the figures have slightly increased over time because of improved detection. [31] This amounts to 1 case per every 50,000 births. [32]
Perforin mutations account for approximately 20% of cases of FEL, with a somewhat higher prevalence (30%) in children of Turkish descent. Chromosome arm 9q mutations account for approximately 10% of familial cases; the remainder of FEL cases are caused by mutations in as yet unidentified genes. [33]
Race-, sex-, and age-related demographics
Hemophagocytic lymphohistiocytosis has not been epidemiologically shown to have a predilection for persons of any race. A sample of European countries, including Sweden, England, and Italy, has reported similar statistical incidences. [19]
The disease has an equal distribution among males and females.
The age of onset is usually in persons younger than 1 year for the familial form but can be later for the secondary sporadic form, usually after age 6 years. [13] Although the familial form of the disease frequently affects infants from birth to age 18 months, familial forms have been reported in individuals as old as 8 years, and adult onset has been reported. At this point, no criteria for age have been established, and an upper age limit does not exist. [32]
Prognosis
Morbidity/mortality
Familial hemophagocytic lymphohistiocytosis is uniformly fatal if not treated; the median survival time reported in various studies is 2-6 months after diagnosis. The historical series collected by the International Hemophagocytic Lymphohistiocytosis Registry reports a less than 10% probability that the patient survives for 3 years. [34] Even with treatment, only 21-26% can be expected to survive 5 years. Remission is always temporary, as the disease inevitably returns. Bone marrow transplant is the only hope for cure. One study found that 50% of deaths from FEL were due to invasive fungal infections, which are probably underdiagnosed. [32] The outcomes of secondary hemophagocytic lymphohistiocytosis vary.
A study of 122 patients from the International Registry for hemophagocytic lymphohistiocytosis found that the overall estimated 5-year survival rate was 21%, with 66% of patients who received bone marrow transplantation (BMT) surviving 5 years versus only 10.1% of patients treated with chemotherapy alone. [34] More recent studies have shown that the HLH-94 protocol resulted in an overall survival rate of 55%. Success or failure of an allogeneic BMT is the most important long-term prognostic factor. Unfortunately, many cases are diagnosed late in the course of the disease, after irreversible damage has occurred.
Although patients with hemophagocytic lymphohistiocytosis are at high risk for death early in their disease course, steroids, intravenous immunoglobulin (IVIG), or both may be sufficient as first-line therapy for selected patients. [35]
The ratio of C16-ceramide to sphingosine was elevated in those who died despite appropriate treatment, but remained low in survivors, implying that this ratio may be of prognostic significance. [36] The balance of ceramide and sphingosine may be pivotal in the clinical outcome for these patients.
Complications
Complications of individual drugs in the treatment regimen are outlined in the Medication section.
Complications due to a subsequent transplant are numerous and include both acute and chronic graft versus host disease, acute inflammatory events, respiratory distress syndrome, and exacerbation of neurologic symptoms. [37]
Patient Education
All physicians who treat young patients must be aware of life-threatening diseases such as hemophagocytic lymphohistiocytosis. Pediatricians, dermatologists, and neurologists should especially take note because the presenting symptoms of hemophagocytic lymphohistiocytosis are likely to bring the patient into their offices. Any suspicion warrants a referral to a pediatric hematologic-oncologist who is equipped with the necessary tools to make a rapid diagnosis.
-
A lymph node biopsy is performed. Note that a marking pen has been used to outline the node before removal and that a silk suture has been used to provide traction to assist the removal.




