Practice Essentials
Epidermolysis bullosa (EB) is a rare group of inherited disorders that manifests as blistering or erosion of the skin and, in some cases, the epithelial lining of other organs, in response to little or no apparent trauma. See the image below.
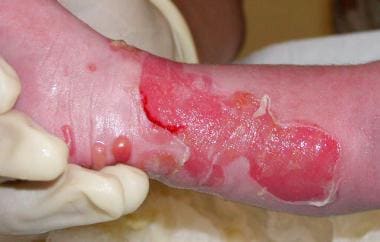 Ruptured bulla and newly erupted bulla of the leg in a newborn with epidermolysis bullosa simplex (EBS).
Ruptured bulla and newly erupted bulla of the leg in a newborn with epidermolysis bullosa simplex (EBS).
Signs and symptoms
Perform a complete physical examination with emphasis on inspecting all skin areas and mucosal surfaces. Evaluate the size, location, and character of the blisters and determine the level at which the blister forms.
Examine the patient for involvement of the nails, hair, or teeth.
Areas prone to blistering due to pressure, trauma, or excessive heating include the fingers, hands, elbows, feet, legs, and diaper area (in infants). See the images below.
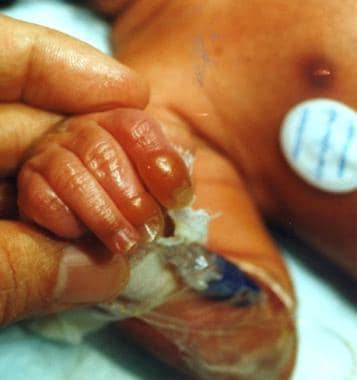 Junctional epidermolysis bullosa (JEB). Image shows a newborn with bulla of the finger, the usual site of blistering secondary to trauma.
Junctional epidermolysis bullosa (JEB). Image shows a newborn with bulla of the finger, the usual site of blistering secondary to trauma.
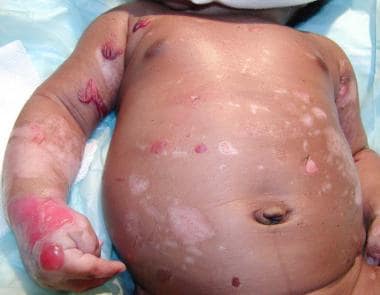
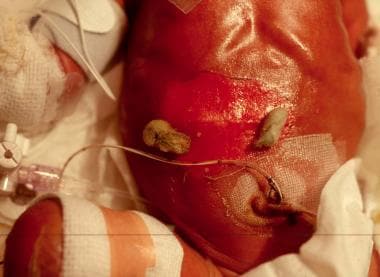 Ruptured bullae on the abdomen in a neonate with junctional epidermolysis bullosa (JEB).
Ruptured bullae on the abdomen in a neonate with junctional epidermolysis bullosa (JEB).
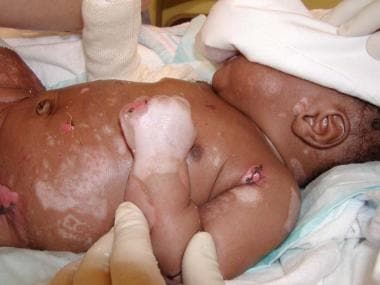
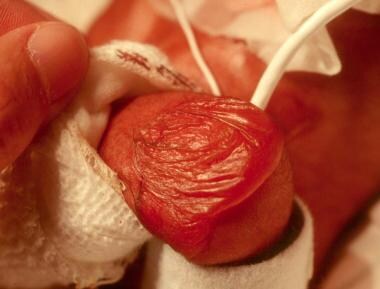 A large bullae on the elbow in a neonate with junctional epidermolysis bullosa (JEB).
A large bullae on the elbow in a neonate with junctional epidermolysis bullosa (JEB).
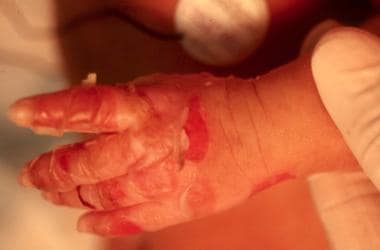 Ruptured bulla of the hand in a newborn with epidermolysis bullosa simplex (EBS).
Ruptured bulla of the hand in a newborn with epidermolysis bullosa simplex (EBS).
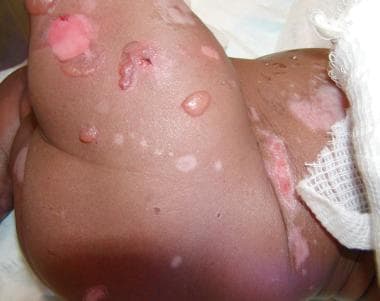
Congenital localized absence of skin is now known to be a phenotypic pattern that neonates with any major form of epidermolysis bullosa may demonstrate at birth. See the images below.
 Junctional epidermolysis bullosa (JEB) with an associated defect of a congenital absence of the skin.
Junctional epidermolysis bullosa (JEB) with an associated defect of a congenital absence of the skin.
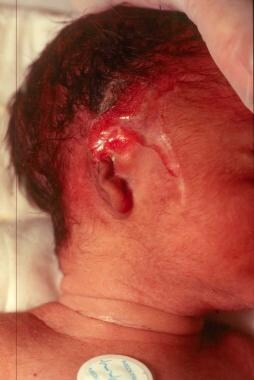 Junctional epidermolysis bullosa (JEB) with an associated defect of a congenital absence of the skin and an ear anomaly.
Junctional epidermolysis bullosa (JEB) with an associated defect of a congenital absence of the skin and an ear anomaly.
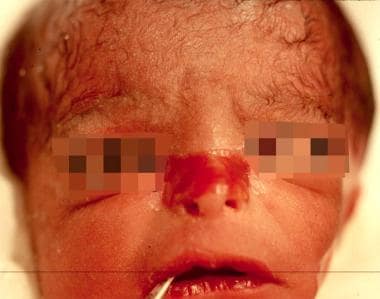 Congenital localized absence of skin, nose, in a neonate with junctional epidermolysis bullosa (JEB).
Congenital localized absence of skin, nose, in a neonate with junctional epidermolysis bullosa (JEB).
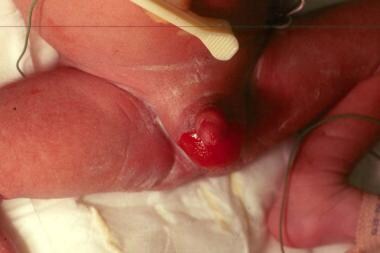 Congenital localized absence of skin on the scrotum in a neonate with junctional epidermolysis bullosa (JEB). Absence of the foreskin was also noted.
Congenital localized absence of skin on the scrotum in a neonate with junctional epidermolysis bullosa (JEB). Absence of the foreskin was also noted.
Diagnostics
Also see Workup.
Transmission electron microscopy
Electron microscopy determines the level of skin cleavage in epidermolysis bullosa (EB) and permits visualization and semiquantitative assessment of specific structures, which are known to be altered in selected epidermolysis bullosa subtypes. See the image below.
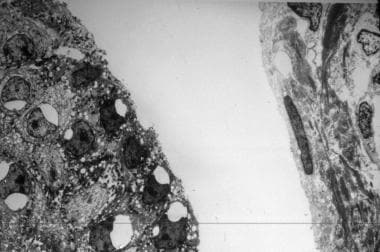 Electron micrograph of a skin sample shows cleavage in the intralaminar lucida in a neonate with junctional epidermolysis bullosa (JEB).
Electron micrograph of a skin sample shows cleavage in the intralaminar lucida in a neonate with junctional epidermolysis bullosa (JEB).
Very few highly proficient electron microscopy laboratories are now available worldwide.
Immunofluorescence mapping (IFM)
IFM is as diagnostically reliable as transmission electron microscopy for several type of epidermolysis bullosa. IFM, when coupled with the use of specific monoclonal antibodies, can provide considerable insight into not only the major type of epidermolysis bullosa but also into the structural protein most likely mutated.
Mutation analysis
Mutation analysis is the ultimate means of determining the mode of inheritance and the precise site and type of molecular mutation. It is the recommended technique; prenatal and preimplantation diagnosis can be performed. It is very labor intensive and expensive to perform. Currently, only a few research or commercial laboratories are equipped to perform this analysis.
Management
Please see Treatment for a full discussion.
The treatment of epidermolysis bullosa (EB) is primarily preventive and supportive. Once blistering has occurred, the blister should be punctured with a sterile needle or a blade. This may prevent the accumulation of fluid and pressure and may thus prevent the blister from extending. Complete and gentle drainage of the fluid, accomplished by leaving the roof of the blister intact and by covering the affected area with white petrolatum–impregnated gauze, helps to promote an environment most optimal for healing. If the blister repeatedly refills with fluid, it should be drained several times.
In May 2023, the US Food and Drug Administration approved Vyjuvek (beremagene geperpavec), the first topical gene therapy drug for the treatment of wounds in dystrophic epidermolysis bullosa, in patients aged 6 months and older.
A topical gel, Filsuvez (birch triterpenes), was approved by the FDA in December 2023 for treatment of wounds in patients aged 6 months and older with junctional and dystrophic epidermolysis bullosa.
Background
More than 30 types of epidermolysis bullosa have been described, rendering categorization of the types controversial and often confusing. An international consensus meeting in Vienna, Austria in 2008 reaffirmed the following currently used names for the four major types of epidermolysis bullosa [1] :
-
Epidermolytic - Epidermolysis bullosa simplex (EBS)
-
Lucidolytic - Junctional epidermolysis bullosa (JEB)
-
Dermolytic - Dystrophic epidermolysis bullosa (DEB)
-
Multiple levels of blistering - Kindler syndrome
These major types were based on the precise ultrastructural level at which the split responsible for blistering occurs. The leading authority on epidermolysis bullosa added Kindler syndrome as the fourth major epidermolysis bullosa type in 2008 with a unique clinical phenotype—photosensitivity. [1]
The three main types of epidermolysis bullosa were clinically and histologically delineated by the 1960s. [2] In the 1970s, electron microscopy revealed abnormal epidermal keratin filaments in epidermolysis bullosa simplex, disordered dermal anchoring fibrils in dystrophic epidermolysis bullosa, and defective hemidesmosomes in junctional epidermolysis bullosa. Antigens identified with immunohistochemistry in the 1980s [3, 4] led to discovery of the major epidermolysis bullosa genes in the 1990s. [5, 6, 7]
The identified genes include those that encode keratins 5 and 14 in epidermolysis bullosa simplex, collagen VII in dystrophic epidermolysis bullosa, and laminin 5 in Herlitz junctional epidermolysis bullosa. Toward the end of millennium, as the complex structure of desmosomes and hemidesmosomes was unraveled, the genes responsible for the rare subtypes were found, including those that encode α6β4 integrin in epidermolysis bullosa with pyloric atresia, plectin in epidermolysis bullosa with muscular dystrophy, and plakophilin in epidermolysis bullosa with ectodermal dysplasia.
During the past few years, systemic data collection and analysis have been performed on several thousands of patients with epidermolysis bullosa worldwide, and more than 1000 mutations, encompassing more than 10 structural genes, have now been documented. More has been learned about the molecular basis of epidermolysis bullosa, a group of diseases that shares clinical or molecular features with several other genodermatoses.
Eponyms associated with different forms of epidermolysis bullosa include the following:
-
Dowling-Meara
-
Köebner
-
Weber-Cockayne
-
Kallin
-
Mendes de Costa
-
Herlitz
-
Ogna
-
Carmi
-
Cockayne-Touraine
-
Pasini
-
Hallopeau-Siemens
-
Shabbir
-
Laryngoonychocutaneous (LOC)
Pathophysiology
Cytolysis causes blisters in the epidermis or basement membrane zone of the skin. In epidermolysis bullosa simplex, cytolysis causes blisters in the basal or spinous layers of the epidermis, and keratinocytes often have abnormal density and organization of keratin filaments. In junctional epidermolysis bullosa, the epidermis separates from the basal lamina, forming a blister cavity in the plane of the lamina lucida, where hemidesmosome structure and density are frequently diminished. In dystrophic epidermolysis bullosa, the basal lamina remains attached to the epidermis, but the blister cavity forms beneath the lamina densa of dermoepidermal junction, and anchoring fibrils may appear abnormal, reduced in number, or altogether absent.
Epidemiology
Frequency
United States
The exact prevalence of epidermolysis bullosa is unknown. Mild variants have been estimated to occur as frequently as 1 per 50,000 births. The more severe varieties are believed to occur in 1 per 500,000 births annually.
Between 1986 and 2002, the National Institutes of Health (NIH) funded the National EB Registry (NEBR), a cross-sectional and longitudinal epidemiological study of epidermolysis bullosa patients across the entire continental United States. Nearly 3300 epidermolysis bullosa patients were identified, enrolled, classified, clinically characterized, and followed for outcomes. The incidence and prevalence of epidermolysis bullosa was estimated at approximately 19 cases per 1 million live births and 11 cases per 1 million population. [8, 9] These data were then used to estimate carrier frequencies for epidermolysis bullosa within the United States, which is between 1 in 300 and 1 in 800. [10]
The prevalence of inherited epidermolysis bullosa subtypes in the United States in January 2002, based on the NEBR was as follows [9] :
-
Epidermolysis bullosa simplex - 6 cases per 1 million population
-
Junctional epidermolysis bullosa - 0.49 case per 1 million population
-
Dominant dystrophic epidermolysis bullosa - 1.49 cases per 1 million population
-
Recessive dystrophic epidermolysis bullosa - 1.35 cases per 1 million population
The incidence of inherited epidermolysis bullosa subtypes in the United States from 1986 to 2002, based on the NEBR was as follows [9] :
-
Epidermolysis bullosa simplex - 7.87 cases per 1 million live births
-
Junctional epidermolysis bullosa - 2.68 per 1 million live births
-
Dominant dystrophic epidermolysis bullosa - 12.12 per 1 million live births
-
Recessive dystrophic epidermolysis bullosa - 3.05 per 1 million live births
International
The point prevalence of all forms of epidermolysis bullosa in Scotland was 49.0 cases per million, comprising epidermolysis bullosa simplex at 28.6 cases per million and dystrophic epidermolysis bullosa at 20.4 cases per million population. [11]
The incidence rate of new cases of epidermolysis bullosa diagnosed per year in Northern Ireland during a 23-year period (1962-84) is 1.4 cases per million and the prevalence of all forms estimated at 32 cases per million population. The prevalence of simplex, junctional, and dystrophic forms is 28 cases, 0.7 case, and 3 cases per million population, respectively. [12]
The estimated prevalence of each type of epidermolysis bullosa in Japan was as follows: simplex type, 2.9-4 cases per million population; junctional type, 0.15-0.20 cases per million population; dominant dystrophic type, 1.1-1.5 cases per million population; and recessive dystrophic type, 1.5-2.1 cases per million population. [13]
The prevalence of epidermolysis bullosa in Croatia, Yugoslavia, from 1960-1987 was 9.5 cases per million population. [14]
In the United Kingdom, prevalence rates of epidermolysis bullosa have been estimated to range from 15-32 cases per million population. [11, 12, 15]
Data from the Australasian EB Registry provided a prevalence estimate of 10 cases per million live births. [16]
Race
The epidermolysis bullosa simplex Ogna variant has been described in Norwegian individuals.
Sex
Epidermolysis bullosa is an autosomal inherited disorder. The incidence does not differ by sex.
Age
The onset of epidermolysis bullosa simplex is at birth or early infancy. The onset of junctional epidermolysis bullosa is at birth. The onset of dystrophic epidermolysis bullosa is at birth or early childhood. The onset of Kindler syndrome is usually within the first year of life.
Prognosis
Epidermolysis bullosa is chronic. Patients should restrict and modify their activity to avoid the serious complications of blistering. Depending on the type of epidermolysis bullosa, disease severity may range from occasional mild blistering of the hands and feet to severe and widespread formation of bullae. These lesions may result in nonhealing erosions, infection, scarring, and joint contracture. Mortality is also related to the abnormalities or anomalies associated with epidermolysis bullosa.
Epidermolysis bullosa continues to be devastating disease with high incidence of aggressive squamous cell carcinoma (SCC). SCC is the most serious complication of epidermolysis bullosa within adults, especially those with Hallopeau-Siemens recessive dystrophic epidermolysis bullosa. By mid adulthood, nearly all patients have had at least one SCC, and nearly 80% die from metastatic SCC. [17] recessive dystrophic epidermolysis bullosa SCC is highly aggressive and has early metastatic spread. Recessive dystrophic epidermolysis bullosa SCC most commonly occurs in chronic nonhealing ulcers and over bony prominences on limbs where UV light exposure is minimal. Unlike most cancers, recessive dystrophic epidermolysis bullosa SCC is generally well-differentiated but behaves in a highly aggressive manner and has high metastatic potential. [18]
Risk of death from renal disease is noted. Causes include renal failure, poststreptococcal glomerulonephritis, secondary amyloidosis, and chronic mechanical obstruction. The cumulative risk of death from renal failure among patients with Hallopeau-Siemens recessive dystrophic epidermolysis bullosa is 12.3% by age 35 years. [19] Death may occur during infancy or early childhood, due to sepsis, renal failure, upper airway occlusion, or failure to thrive. [19, 20]
Healing of dystrophic epidermolysis bullosa results in dystrophic or scarring change. In epidermolysis bullosa simplex, when blisters cleave in the epidermis, healing occurs without scarring. In junctional epidermolysis bullosa, when blisters cleave below the epidermis but above the basal lamina, blistering leads to mild atrophic changes.
-
Ruptured bulla and newly erupted bulla of the leg in a newborn with epidermolysis bullosa simplex (EBS).
-
Dystrophic epidermolysis bullosa (DEB) with multiple blisters and erosions.
-
Dystrophic epidermolysis bullosa (DEB) with generalized blistering and erosion.
-
Dystrophic epidermolysis bullosa (DEB) that subsequently healed, with scarring.
-
Junctional epidermolysis bullosa (JEB). Image shows a newborn with bulla of the finger, the usual site of blistering secondary to trauma.
-
Junctional epidermolysis bullosa (JEB) with an associated defect of a congenital absence of the skin.
-
Junctional epidermolysis bullosa (JEB) with an associated defect of a congenital absence of the skin and an ear anomaly.
-
Contrast-enhanced radiograph of the abdomen suggestive of pyloric atresia (PA) in a patient with junctional epidermolysis bullosa (JEB). The association between PA and EB is a distinct entity and is now referred to as PA-EB syndrome.
-
Electron micrograph of a skin sample shows cleavage in the intralaminar lucida in a neonate with junctional epidermolysis bullosa (JEB).
-
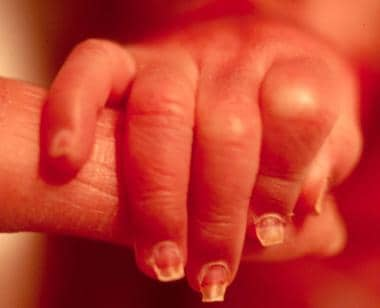
Dystrophic nails in a neonate with junctional epidermolysis bullosa (EB).
-
Congenital localized absence of skin, nose, in a neonate with junctional epidermolysis bullosa (JEB).
-
Ruptured bullae on the abdomen in a neonate with junctional epidermolysis bullosa (JEB).
-
A large bullae on the elbow in a neonate with junctional epidermolysis bullosa (JEB).
-
Congenital localized absence of skin on the scrotum in a neonate with junctional epidermolysis bullosa (JEB). Absence of the foreskin was also noted.
-
Severe right hydronephrosis in a neonate with junctional epidermolysis bullosa (JEB).
-
Genitourinary obstruction in a fetus with junctional epidermolysis bullosa (JEB). Prenatal ultrasonography reveals severe right hydronephrosis.
-
Ruptured bulla of the hand in a newborn with epidermolysis bullosa simplex (EBS).
-
Abdominal radiography revealing a single gastric bubble in a neonate with pyloric atresia (PA) and junctional epidermolysis bullosa (JEB).
Tables
- Table 1. Morphologic and molecular features of Major Types and Subtypes of Epidermolysis Bullosa
- Table 2. Major Epidermolysis Bullosa Types and Subtypes
- Table 3. Mutational Analyses and Inherited Epidermolysis Bullosa [24, 25, 26]
- Table 4. Major Epidermolysis Bullosa Subtypes and Their Targeted Proteins (2008 International Consensus Report [27] )
Type or Subtype |
Level of Blister Formation |
Protein Affected and Immunofluorescence Staining Pattern |
Epidermolysis bullosa simplex (EBS) |
||
Suprabasal |
Suprabasal epidermis |
Transglutaminase 5: Normal, reduced, or absent |
|
|
Desmoplakin: Reduced or absent |
|
|
Plakoglobin: Reduced or absent |
|
|
Plakophilin 1: Reduced or absent |
|
Basal epidermis |
Keratin 5 or keratin 14: Usually normal |
|
|
Exophilin 5: Absent |
|
|
Plectin: Reduced or absent |
|
|
Bullous pemphigoid antigen-1: Absent |
Junctional epidermolysis bullosa (JEB) |
||
JEB, generalized severe |
Intralamina lucida |
Laminin-332: Absent or markedly reduced |
JEB, generalized intermediate |
Intralamina lucida |
Laminin-332: Reduced |
JEB with pyloric atresia |
Intralamina lucida |
Type XVII collagen: Reduced or absent |
|
Intralamina lucida |
α6β4 integrin: Absent or markedly reduced |
JEB, late onset |
Intralamina lucida |
Type XVII collagen: Reduced or abnormal pattern |
JEB with respiratory and renal involvement |
Intralamina lucida |
α6β4 integrin: Absent or normal |
JEB, localized |
Intralamina lucida |
Type XVII collagen: Reduced or absent |
|
|
α6β4 integrin: Reduced |
|
|
Laminin-332: Reduced |
JEB-inversa |
Intralamina lucida |
Laminin-332: Reduced |
JEB-LOC syndrome (LOC, laryngo-onycho-cutaneous) |
No blistering |
Laminin-332: Normal |
Dominant dystrophic epidermolysis bullosa (DDEB) |
||
All subtypes |
Sublamina densa (dermal) |
Type XVII collagen: normal or reduced |
Bullous dermolysis of the newborn |
Sublamina densa (dermal) |
Type XVII collagen: Granular staining within basal keatinocytes; reduced/absent staining along dermoepidermal junction during active disease; normal staining along dermoepidermal junction during inactive disease |
Recessive dystrophic epidermolysis bullosa (RDEB) |
||
Bullous dermolysis of the newborn |
Sublamina densa (dermal) |
Type XVII collagen: Granular staining within basal keatinocytes; reduced/absent staining along dermoepidermal junction during active disease; normal staining along dermoepidermal junction during inactive disease |
Generalized severe |
Sublamina densa (dermal) |
Type XVII collagen: Absent or markedly reduced |
Generalized intermediate |
Sublamina densa (dermal) |
Type XVII collagen: Reduced |
Localized |
Sublamina densa (dermal) |
Type XVII collagen: Normal or reduced or normal |
All other subtypes |
Sublamina densa (dermal) |
Type XVII collagen: Reduced |
Level of Skin Cleavage |
Major Type |
Known Targeted Protein |
Intraepidermal |
Suprabasal epidermolysis bullosa simplex |
Transglutaminase 5; plakophillin 1 desmoplakin; plakoglobin |
|
Basal epidermolysis bullosa simplex |
Keratins 5 and 14; plectin; exophilin 5(Slac2-b); bullous pemphigoid antigen1 |
Intralamina lucida |
Junctional epidermolysis bullosa, generalized |
Laminin-332; type XVII collagen; α6β4 integrin; α3 integrin |
|
Junctional epidermolysis bullosa, localized |
Type XVII collagen; laminin-332; α6β4 integrin |
Sublamina densa |
Dominant dystrophic epidermolysis bullosa |
Type VII collagen |
|
Recessive dystrophic epidermolysis bullosa |
Type VII collagen |
Mixed |
Kindler syndrome |
Kindlin-1 |
Epidermolysis Bullosa Subtype |
Target Gene (Protein) |
Types of Known Mutations |
Epidermolysis bullosa simplex (EBS) - Suprabasal |
PKP1 (plakophilin1) |
Splice site, nonsense, deletion, deletion/insertion, insertion |
|
DSP (desmoplakin) |
Nonsense, deletion, missense |
|
TGMS |
Missense, deletion, deletion/insertion |
|
JUP |
Nonsense, splice site |
EBS - Basal |
KRT5 (keratin-5) |
Missense, deletion, splice site, nonsense, deletion/insertion |
|
KRT14 (keratin-14) |
Missense, deletion, nonsense, splice site, deletion/insertion, insertion |
|
PLEC (plectin) |
Nonsense, deletion, insertion, deletion/insertion, splice site, missense, |
|
EXPH5 |
Deletion, nonsense, insertion |
|
DST |
Nonsense |
Junctional epidermolysis bullosa (JEB) - Generalized |
LAMA3 |
Nonsense, deletion, splice site |
|
LAMB3 |
Nonsense, deletion, splice site, insertion, |
|
LAMC2 |
Nonsense, deletion, splice site, deletion/insertion |
JEB, generalized/localized |
LAMA3 |
Missense, nonsense, insertion, splice site |
|
LAMB3 |
Missense, nonsense, splice site, deletion, insertion, deletion/insertion |
|
LAMC2 |
Nonsense, deletion, deletion/insertion, insertion, splice site |
|
COL17A1 (type XVII collagen) |
Nonsense, deletion, splice site, insertion, missense |
|
ITGB4 (α6β4 integrin) |
Deletion, splice site, missense |
JEB, late onset |
COL17A1 (type XVII collagen) |
Missense |
JEB with pyloric atresia |
ITGB4 (α6β4 integrin)
|
Nonsense, missense, deletion, splice site, insertion deletion/insertion |
|
ITGA6 |
Deletion, missense, nonsense, splice site |
JEB with pyloric atresia |
ITGA3 |
Missense, deletion, splice site |
JEB with respiratory and renal involvement |
LAMA3A |
Insertion, nonsense |
JEB, severe generalized |
COL17A1 (type VII collagen) |
Nonsense, deletion, splice site, insertion, deletion/insertion, missense, |
Dystrophic epidermolysis bullosa, generalized and localized |
COL17A1 (type VII collagen) |
Missense, nonsense, deletion, insertion, splice site, deletion/insertion |
Dystrophic epidermolysis bullosa (all subtypes) |
COL17A1 (type VII collagen) |
Missense, splice site, deletion |
Kindler syndrome |
KIND1 (kindling-1) |
Nonsense, deletion, splice site, insertion, deletion/insertion |
Major Epidermolysis Bullosa Type |
Major Epidermolysis Bullosa Subtypes |
Targeted Protein(s) |
Epidermolysis bullosa simplex (EBS) |
Suprabasal subtypes |
|
|
Acantholytic EBS (EBS-acanth) |
Desmoplakin, plakoglobin |
|
Acral peeling skin syndrome (APSS) |
Transglutaminase 5 |
|
EBS superficialis (EBSS) |
Unknown |
|
Plakophilin-1 deficiency |
Plakophilin-1 |
|
Plakoglobin deficiency (EBS-plakoglobin) |
Plakoglobin |
|
Desmoplakin deficiency (EBS-desmoplakin) |
Desmoplakin |
|
Basal subtypes |
|
|
EBS, localized (EBS-loc) |
K5, K14 |
|
EBS, generalized severe (EBS-gen sev) |
K5, K14 |
|
EBS, generalized intermediate (EBS-gen intermed) |
K5, K14 |
|
EBS with mottled pigmentation (EBS-MP) |
K5 |
|
EBS with migratory circinate (EBS-migr) |
Plectin |
|
EBS with pyloric atresia (EBS-PA) |
Plectin; α6β4 integrin |
|
EBS, autosomal recessive K14 (EBS-AR K14) |
K14 |
|
EBS with muscular dystrophy (EBS-MD) |
Plectin |
|
EBS, Ogna (EBS-Og) |
Plectin |
|
EBS, migratory circinate (EBS-migr) |
K5 |
|
EBS, autosomal recessive-BP230 deficiency (EBS-AR BP230) |
Bullous pemphigoid antigen-1 (BP230) |
|
EBS, autosomal recessive-exophilin 5 deficiency (EBS-AR exophilin 5)BP230 |
Exophilin 5 |
Junctional epidermolysis bullosa (JEB) |
JEB, generalized severe (JEB-gen sev) |
laminin-332 |
|
JEB, generalized intermediate (JEB-gen intermed) |
laminin-332; type XVII collagen |
|
JEB late onset (JEB-LO) |
type XVII collagen |
|
JEB with pyloric atresia (JEB-PA) |
α6β4 integrin |
|
JEB, with respiratory and renal involvement (JEB-RR) |
α3 integrin |
|
JEB localized (JEB-loc) |
type VII collagen, α6β4 integrin, laminin-332 |
|
JEB, inversa (JEB-inv; JEB-I) |
laminin-332 |
|
JEB-LOC syndrome |
laminin-332, isoform α3 chain |
Dominant dystrophic epidermolysis bullosa (DDEB) |
DDEB, generalized (DDEB-gen) |
type VII collagen |
|
DDEB, acral (DDEB-ac) |
type VII collagen |
|
DDEB, pretibial (DDEB-Pt) |
type VII collagen |
|
DDEB, pruriginosa (DDEB-Pr) |
type VII collagen |
|
DDEB, nails only (DDEB-na) |
type VII collagen |
|
DDEB, bullous dermolysis of newborn (DDEB-BDN) |
type VII collagen |
Recessive dystrophic epidermolysis bullosa (RDEB) |
RDEB, severe generalized (RDEB-sev gen) |
type VII collagen |
|
RDEB, generalized other (RDEB, generalized mitis (RDEB-O) |
type VII collagen |
|
RDEB, inversa (RDEB-I) |
type VII collagen |
|
RDEB, pretibial (RDEB-Pt) |
type VII collagen |
|
RDEB, pruriginosa (RDEB-Pr) |
type VII collagen |
|
RDEB, centripetalis (RDEB-Ce) |
type VII collagen |
|
RDEB, bullous dermolysis of newborn (RDEB-BDN) |
type VII collagen |
Kindler syndrome |
|
kindlin-1 |





