Practice Essentials
Skeletal dysplasias, also known as osteochondrodysplasias, are a heterogeneous group of heritable disorders characterized by abnormalities of cartilage and bone growth, resulting in abnormal shape and size of the skeleton and disproportion of the long bones, spine, and head. They differ in natural histories, prognoses, inheritance patterns, and etiopathogenetic mechanisms. Typified by short stature (defined as height that is three or more standard deviations below the mean height for age), skeletal dysplasias can be accompanied by involvement of other systems, including the neurologic, respiratory, and cardiac systems.
The molecular basis for a large majority of these disorders in now known. Commonly seen skeletal dysplasias include achondroplasia, osteogenesis imperfecta, thanatophoric dysplasia, campomelic dysplasia, and hypochondroplasia. (See the images below.)
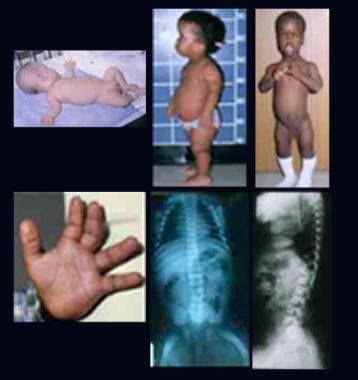 Infant and 2 children with achondroplasia. Note relatively normal-sized trunk, a large head, rhizomelic shortening of the limbs, lumbar lordosis, and trident hands. Radiographs demonstrate abnormal pelvis with small square iliac wings, horizontal acetabular roofs, and narrowing of the greater sciatic notch, an oval translucent area at the proximal ends of the femora, caudal narrowing of the interpedicular distances in the lumbar region, short pedicles, and lumbar lordosis.
Infant and 2 children with achondroplasia. Note relatively normal-sized trunk, a large head, rhizomelic shortening of the limbs, lumbar lordosis, and trident hands. Radiographs demonstrate abnormal pelvis with small square iliac wings, horizontal acetabular roofs, and narrowing of the greater sciatic notch, an oval translucent area at the proximal ends of the femora, caudal narrowing of the interpedicular distances in the lumbar region, short pedicles, and lumbar lordosis.
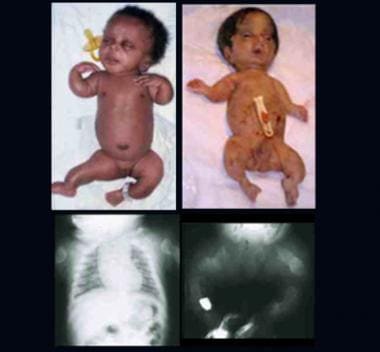 Two infants with perinatal lethal form of osteogenesis imperfecta. Note short-limbed skeletal dysplasia, deformed extremities, and relatively large head. Radiographs show short, thick, ribbonlike long bones with multiple fractures and callus formation at all sites (ribs, long bones).
Two infants with perinatal lethal form of osteogenesis imperfecta. Note short-limbed skeletal dysplasia, deformed extremities, and relatively large head. Radiographs show short, thick, ribbonlike long bones with multiple fractures and callus formation at all sites (ribs, long bones).
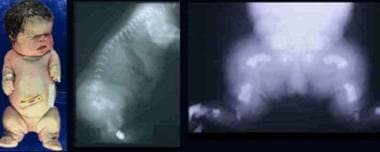 Infant with thanatophoric dysplasia. Note short-limbed dysplasia, large head, short neck, narrow thorax, short and small fingers, and bowed extremities. Radiographs demonstrate thin flattened vertebrae, short ribs, small sacrosciatic notch, extremely short long tubular bones, and markedly short and curved femora (telephone receiver–like appearance).
Infant with thanatophoric dysplasia. Note short-limbed dysplasia, large head, short neck, narrow thorax, short and small fingers, and bowed extremities. Radiographs demonstrate thin flattened vertebrae, short ribs, small sacrosciatic notch, extremely short long tubular bones, and markedly short and curved femora (telephone receiver–like appearance).
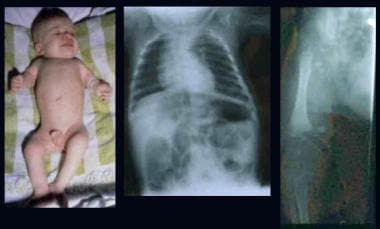 Infant with rhizomelic form of chondrodysplasia punctata (left). Note rhizomelic shortening of limbs, disproportionately short stature, enlarged joints, and contractures. Radiographs depict epiphyseal stipplings on the proximal humerus, both ends of the femora, and lower spine.
Infant with rhizomelic form of chondrodysplasia punctata (left). Note rhizomelic shortening of limbs, disproportionately short stature, enlarged joints, and contractures. Radiographs depict epiphyseal stipplings on the proximal humerus, both ends of the femora, and lower spine.
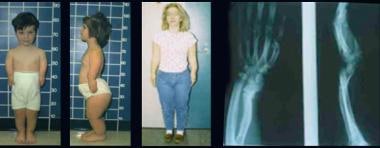 Brother and sister with mesomelic dysplasia (homozygous dyschondrosteosis gene) and a woman with Leri-Weill syndrome. Note disproportionately short stature with mesomelic shortening and deformities of forearms and legs (in mesomelic dysplasia) and short forearms with Madelung-type deformity (in Leri-Weill syndrome).
Brother and sister with mesomelic dysplasia (homozygous dyschondrosteosis gene) and a woman with Leri-Weill syndrome. Note disproportionately short stature with mesomelic shortening and deformities of forearms and legs (in mesomelic dysplasia) and short forearms with Madelung-type deformity (in Leri-Weill syndrome).
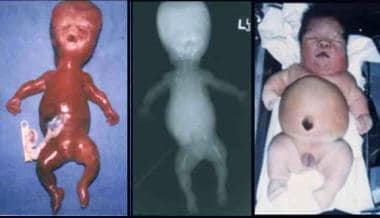 Infant with Beemer-type (left) and an infant with Majewski-type (right) short-rib syndrome (SRS). Note severe micrognathia/retrognathia with cleft palate, apparently low-set and malformed ears, small and narrow chest, protuberant abdomen with omphalocele, and short and slightly curved limbs with bilateral postaxial polydactyly (Beemer-type SRS), a large head, short nose, flat nasal bridge, central cleft of upper and lower lips, short neck, short chest, protuberant abdomen, abdomen, ambiguous genitalia, short limbs, and preaxial and postaxial polydactyly (Majewski-type SRS).
Infant with Beemer-type (left) and an infant with Majewski-type (right) short-rib syndrome (SRS). Note severe micrognathia/retrognathia with cleft palate, apparently low-set and malformed ears, small and narrow chest, protuberant abdomen with omphalocele, and short and slightly curved limbs with bilateral postaxial polydactyly (Beemer-type SRS), a large head, short nose, flat nasal bridge, central cleft of upper and lower lips, short neck, short chest, protuberant abdomen, abdomen, ambiguous genitalia, short limbs, and preaxial and postaxial polydactyly (Majewski-type SRS).
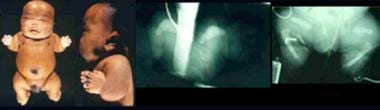 Infant with atelosteogenesis. Note short-limbed dysplasia, relative macrocephaly, and short neck. Radiographs demonstrate boomeranglike triangular or oval form of the long bones (humeri), absent radii, markedly delayed ossification of phalanges, short femora, and absent fibulae.
Infant with atelosteogenesis. Note short-limbed dysplasia, relative macrocephaly, and short neck. Radiographs demonstrate boomeranglike triangular or oval form of the long bones (humeri), absent radii, markedly delayed ossification of phalanges, short femora, and absent fibulae.
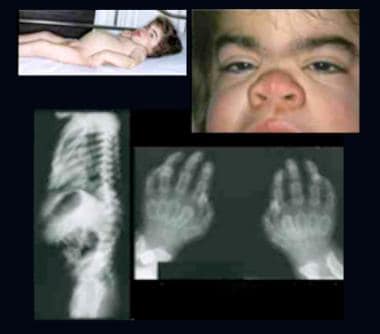 Child with Hurler syndrome (mucopolysaccharidosis type IH). Note dysplasia, scaphocephalic macrocephaly, coarse facial features, depressed nasal bridge, broad nasal tip, thick lips, short neck, protuberant abdomen, inguinal hernia, joint contractures, and claw hands. Radiographs demonstrate hook-shaped deformity (anterior wedging) of the L1 and L2 vertebrae; abnormally short, wide, and deformed tubular bones (bullet-shaped) of the hands; and narrow base of the second-to-fifth metacarpals. The distal articular surfaces of the ulna and radius are slanted toward each other.
Child with Hurler syndrome (mucopolysaccharidosis type IH). Note dysplasia, scaphocephalic macrocephaly, coarse facial features, depressed nasal bridge, broad nasal tip, thick lips, short neck, protuberant abdomen, inguinal hernia, joint contractures, and claw hands. Radiographs demonstrate hook-shaped deformity (anterior wedging) of the L1 and L2 vertebrae; abnormally short, wide, and deformed tubular bones (bullet-shaped) of the hands; and narrow base of the second-to-fifth metacarpals. The distal articular surfaces of the ulna and radius are slanted toward each other.
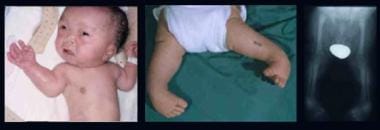 Infant with Larsen syndrome. Note the flat face with depressed nasal bridge, prominent forehead, hypertelorism, cleft palate, talipes equinovarus, and dislocations of elbows, hips, and knees. Radiograph demonstrates dislocation at the knee.
Infant with Larsen syndrome. Note the flat face with depressed nasal bridge, prominent forehead, hypertelorism, cleft palate, talipes equinovarus, and dislocations of elbows, hips, and knees. Radiograph demonstrates dislocation at the knee.
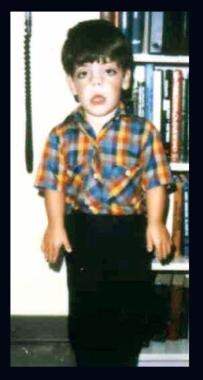 Child with Robinow syndrome. Note moderate short stature, flat facial profile (fetal face–like appearance), short forearms, and small hands.
Child with Robinow syndrome. Note moderate short stature, flat facial profile (fetal face–like appearance), short forearms, and small hands.
During the 1950s and 1970s, many new bone dysplasias were identified based on clinical manifestations, radiographic findings, inheritance patterns, and morphology of the growth plate. In the 1980s, research focused on defining the natural history and variability of the disorders. In the 1990s, the focus shifted toward elucidating the responsible mutations and characterizing the pathogenetic mechanisms by which the mutations disrupt bone growth.
In 1997, the International Working Group on Bone Dysplasias proposed a newly revised "International Nomenclature and Classification of the Osteochondrodysplasias." [1] In the revised nomenclature, families of disorders were rearranged based on recent etiopathogenetic information concerning the gene and/or protein defect involved. Disorders for which the basic defect was well documented were regrouped into distinct families in which component disorders result from mutations of the identical gene.
Over the past decades, substantial advances have been made in understanding the underlying genetic abnormalities responsible for most skeletal dysplasias. [2] The 2015 revision of Nosology and Classification of Genetic Skeletal Disorders recognizes 436 genetic skeletal diseases, stemming from mutations in 364 genes, with the disorders organized into 42 groups. [3] Classifications based on the underlying molecular genetic cause are regularly updated. [4, 5, 3, 6]
Skeletal dysplasias can be inherited as autosomal dominant, autosomal recessive, X-linked, or Y-linked. Examples include the following:
-
Autosomal dominant - Fibroblast growth factor 3 ( FGFR3) disorders (achondroplasia, thanatophoric dysplasia, hypochondroplasia) and type II collagen disorders (achondrogenesis II, spondyloepiphyseal dysplasia congenita, Kniest dysplasia)
-
Autosomal recessive - Cartilage-hair hypoplasia, Ellis-van Creveld syndrome, hypophosphatasia, osteopetrosis
-
X-linked dominant - Chondrodysplasia punctata (CDP)
-
X-linked recessive - Conradi-Hunermann CDP
A brief overview of the most common skeletal dysplasias is included below.
Achondroplasia
This is the most common nonlethal skeletal dysplasia, occurring in 1:26,000-1:28,000 live births and affecting 250,000 individuals worldwide. [7, 8]
The molecular mechanism for achondroplasia is a G380R mutation in the FGFR3 transmembrane domain. A gain-of-function mutation, it is present in 99% of affected individuals. Inheritance is autosomal dominant, with 80% of cases involving de novo mutations.
Major characteristics of achondroplasia include the following [7] :
-
Disproportionate short stature with rhizomelic (proximal) shortening of the arms and legs and large head with frontal bossing
-
Trident hand configuration
-
Average final height: 130 cm for men and 125 cm for women
-
Normal intelligence and lifespan
Major complications include the following [9] :
-
Craniocervical junction compression
-
Middle ear infections
-
Obstructive apnea
-
Spinal stenosis
A prospective, multinational, observational study by Savarirayan et al looked at the growth characteristics of children (aged 17 years or below; 363 individuals enrolled) with achondroplasia. All participants in the study were ambulatory and did not need assistance to stand. The mean annualized growth velocities (AGVs) in females and males under age 1 year were 11.6 cm/year and 14.6 cm/year, respectively. A reduction in the AGV was seen by age 1 year in females and males, being 7.1 cm/year and 7.4 cm/year, respectively. A further decrease was found by age 10 years, with a mean AGV of about 3.6 cm/year for both sexes. The mean upper-to-lower body segment ratios for females and males under age 1 year were 2.9 and 2.8, respectively, gradually decreasing in both sexes to approximately 2 by age 4 years. [10]
See also the American Academy of Pediatrics guideline on the care of the child with achondroplasia. [11]
Osteogenesis imperfecta
A heterogenous group of heritable connective tissue disorders, osteogenesis imperfecta (OI) is another common skeletal dysplasia, with a prevalence of 1:15,000-1:20,000 births.
Molecular mechanisms for OI types I-IV are mutations in type 1 collagen genes COLA1 and COLA2. Types V-XII are rarer forms.
Inheritance in OI is as follows:
-
Types I-IV (Sillence classification, 85% of cases): Autosomal dominant
-
Types V-XII: Autosomal recessive, except for type V (autosomal dominant)
Major characteristics of the most common forms of OI are as follows.
All types
All forms are characterized by bone fragility and susceptibility to fracture from minimal trauma.
OI type I - mild form with diagnosis in early childhood
This is characterized by the following:
-
Sclera may be blue
-
Dentinogenesis imperfecta (subtype IB)
-
Normal stature reached
-
Hearing loss in 50% of patients
OI type II - perinatal lethal form
The condition is characterized as follows:
-
Patients may survive the neonatal period
-
Later mortality can occur secondary to pneumonia and respiratory insufficiency
OI type III - progressive deforming form
This form is characterized as follows:
-
Moderate deformity at birth
-
Development of chest wall deformities
-
Most patients are wheelchair dependent
-
Very short stature
-
Variable sclera
-
Dentinogenesis imperfecta and hearing loss are common.
OI type IV - moderately severe form
Characteristics include the following:
-
Mild to moderate bone deformity
-
Variable short stature
-
Hearing loss occurs in some families
-
Variable sclera
Major complications
These include bone fractures, bone deformity, and growth deficiency.
See also Osteogenesis Imperfecta and Genetics of Osteogenesis Imperfecta.
Diagnosis and management of skeletal dysplasias
Diagnosis of skeletal dysplasias requires a multidisciplinary approach and includes radiologic and genetic evaluations. This approach is required for accurate diagnosis and the determination of the best treatment options, as well as for accurate counseling on outcomes and risk of recurrence.
Conventional radiographic examination remains the most useful means of studying the dysplastic skeleton. The skeletal survey should include the skull (anteroposterior [AP], lateral views), chest (AP), spine (AP and lateral views, including dedicated lateral view of the cervical spine), pelvis (AP), tubular bones (AP), and/or hands and feet (AP). [12]
In addition, genetic testing plays a critical role in the diagnosis and management of skeletal dysplasias. Molecular diagnostic techniques have led to the identification of the underlying gene disorders in about two thirds of known skeletal dysplasias.
Treatment is supportive. Medical care for individuals with skeletal dysplasia should be directed at preventing neurologic and orthopedic complications due to spinal cord compression, joint instability, and long bone deformity.
Surgical intervention depends on the signs and symptoms of skeletal dysplasia. For example, progressive kyphosis, which may lead to spinal cord compression and spastic paraparesis, is best treated by anterior and posterior fusion. Lumbar lordosis with spinal stenosis responds to extensive lumbar laminectomy. Surgical decompression is required to relieve edema of the cervicomedullary cord secondary to bony compression.
Pathophysiology
Until skeletal maturity, cartilage persists at the ends of bone in the growth plate, which is responsible for longitudinal bone growth. The cartilaginous template is eventually replaced by bone. Many of the genes mutated in skeletal dysplasias encode proteins that play critical roles in the growth plate. An understanding of the role in growth plate function gives important clues into the molecular pathology of the skeletal dysplasia and makes it easy to understand how a certain mutation causes a particular phenotype. [13] Examples of genes that play a role in growth plate chondrocytes and skeletal dysplasia include the following:
-
Resting zone of the growth plate: SOX9 gene mutation causes camptomelic dysplasia, which is characterized by short and curved bone and is associated with sex reversal in which the female external genitalia does not match the male genotype. A heterozygous mutation is sufficient to cause the disease making this a dominant mutation, despite earlier reports suggesting that camptomelic dysplasia is a recessive disorder.
-
Proliferation zone of the growth plate: FGFR3 gene mutation causes achondroplasia, hypochondroplasia, and thanatophoric dysplasia, despite the variability in severity. [14]
-
Zone of terminal differentiation of the growth plate: RUNX2 gene mutation causes cleidocranial dysplasia. [19]
-
Mutations in type II collagen cause a large number of disorders classified as spondyloepiphyseal dysplasia (ie, spondyloepiphyseal dysplasia congenita, Kniest dysplasia, Stickler syndrome, and achondrogenesis). Mutations in the smaller matrix components, such as type IX collagen and cartilage oligomeric protein, cause multiple epiphyseal dysplasia.
Epidemiology
Frequency
United States
-
Skeletal dysplasias represent approximately 5% of all congenital anomalies.
-
The overall incidence of skeletal dysplasias is approximately 1 case per 4000-5000 births. The true incidence may be twice as high because many skeletal dysplasias do not manifest until short stature, joint symptoms, or other complications arise during childhood.
-
Lethal skeletal dysplasias are estimated to occur in 0.95 per 10,000 deliveries.
-
The 4 most common skeletal dysplasias are thanatophoric dysplasia, achondroplasia, osteogenesis imperfecta, and achondrogenesis. Thanatophoric dysplasia and achondroplasia account for 62% of all lethal skeletal dysplasias.
-
Achondroplasia is the most common nonlethal skeletal dysplasia.
Mortality/Morbidity
See the list below:
-
Among infants with skeletal dysplasias detected at birth, approximately 13% are stillborn, and 44% die during the perinatal period.
-
The overall frequency of skeletal dysplasias in infants who die perinatally is 9.1 per 1000.
Race
See the list below:
-
No racial predilections are described.
Sex
See the list below:
-
Males are primarily affected in X-linked recessive disorders. X-linked dominant disorders may be lethal in males.
-
Otherwise, males and females are usually equally affected by skeletal dysplasias.
Age
See the list below:
-
Skeletal dysplasias are usually detected in the newborn period or during infancy.
-
Some disorders may not manifest until later in childhood.
Prognosis
The prognosis in skeletal dysplasias is variable. Some of these disorders are lethal in the neonatal period, while others present later in childhood with short stature.
-
Infant with rhizomelic form of chondrodysplasia punctata (left). Note rhizomelic shortening of limbs, disproportionately short stature, enlarged joints, and contractures. Radiographs depict epiphyseal stipplings on the proximal humerus, both ends of the femora, and lower spine.
-
Brother and sister with mesomelic dysplasia (homozygous dyschondrosteosis gene) and a woman with Leri-Weill syndrome. Note disproportionately short stature with mesomelic shortening and deformities of forearms and legs (in mesomelic dysplasia) and short forearms with Madelung-type deformity (in Leri-Weill syndrome).
-
Infant with Beemer-type (left) and an infant with Majewski-type (right) short-rib syndrome (SRS). Note severe micrognathia/retrognathia with cleft palate, apparently low-set and malformed ears, small and narrow chest, protuberant abdomen with omphalocele, and short and slightly curved limbs with bilateral postaxial polydactyly (Beemer-type SRS), a large head, short nose, flat nasal bridge, central cleft of upper and lower lips, short neck, short chest, protuberant abdomen, abdomen, ambiguous genitalia, short limbs, and preaxial and postaxial polydactyly (Majewski-type SRS).
-
Infant and 2 children with achondroplasia. Note relatively normal-sized trunk, a large head, rhizomelic shortening of the limbs, lumbar lordosis, and trident hands. Radiographs demonstrate abnormal pelvis with small square iliac wings, horizontal acetabular roofs, and narrowing of the greater sciatic notch, an oval translucent area at the proximal ends of the femora, caudal narrowing of the interpedicular distances in the lumbar region, short pedicles, and lumbar lordosis.
-
Infant with thanatophoric dysplasia. Note short-limbed dysplasia, large head, short neck, narrow thorax, short and small fingers, and bowed extremities. Radiographs demonstrate thin flattened vertebrae, short ribs, small sacrosciatic notch, extremely short long tubular bones, and markedly short and curved femora (telephone receiver–like appearance).
-
Infant with atelosteogenesis. Note short-limbed dysplasia, relative macrocephaly, and short neck. Radiographs demonstrate boomeranglike triangular or oval form of the long bones (humeri), absent radii, markedly delayed ossification of phalanges, short femora, and absent fibulae.
-
Child with Hurler syndrome (mucopolysaccharidosis type IH). Note dysplasia, scaphocephalic macrocephaly, coarse facial features, depressed nasal bridge, broad nasal tip, thick lips, short neck, protuberant abdomen, inguinal hernia, joint contractures, and claw hands. Radiographs demonstrate hook-shaped deformity (anterior wedging) of the L1 and L2 vertebrae; abnormally short, wide, and deformed tubular bones (bullet-shaped) of the hands; and narrow base of the second-to-fifth metacarpals. The distal articular surfaces of the ulna and radius are slanted toward each other.
-
Two infants with perinatal lethal form of osteogenesis imperfecta. Note short-limbed skeletal dysplasia, deformed extremities, and relatively large head. Radiographs show short, thick, ribbonlike long bones with multiple fractures and callus formation at all sites (ribs, long bones).
-
Infant with Larsen syndrome. Note the flat face with depressed nasal bridge, prominent forehead, hypertelorism, cleft palate, talipes equinovarus, and dislocations of elbows, hips, and knees. Radiograph demonstrates dislocation at the knee.
-
Child with Robinow syndrome. Note moderate short stature, flat facial profile (fetal face–like appearance), short forearms, and small hands.








