Practice Essentials
Dubin-Johnson syndrome (DJS) is an inherited, relapsing, benign disorder of bilirubin metabolism. Dubin-Johnson syndrome is characterized by defective bilirubin excretion into bile. This results in reduced hepatic bilirubin clearance. [1] This rare autosomal recessive condition is characterized by conjugated hyperbilirubinemia with normal liver transaminases, a unique pattern of urinary excretion of heme metabolites (coproporphyrins), and the deposition of a pigment that gives the liver a characteristic black color (see the image below).
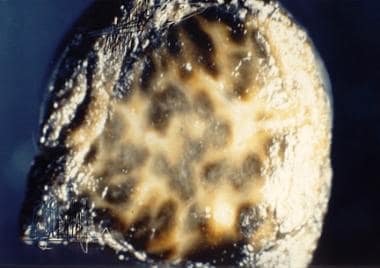 Dubin-Johnson Syndrome. Gross liver specimen from a patient with Dubin-Johnson syndrome showing multiple areas of dark pigmentation. Image courtesy of Cirilo Sotelo-Avila, MD.
Dubin-Johnson Syndrome. Gross liver specimen from a patient with Dubin-Johnson syndrome showing multiple areas of dark pigmentation. Image courtesy of Cirilo Sotelo-Avila, MD.
Signs and symptoms
Patients with Dubin-Johnson syndrome tend to develop nonpruritic jaundice during their teenaged years.
Some patients complain of nonspecific right upper quadrant pain, which has been attributed to the anxiety associated with prolonged diagnostic testing. Hepatosplenomegaly also occurs in some patients, but in most cases, Dubin-Johnson syndrome is asymptomatic.
See Presentation for more detail.
Diagnosis
Laboratory studies reveal conjugated hyperbilirubinemia, with total bilirubin serum levels usually in the 2- to 5-mg/dL range (but potentially as high as 25 mg/dL).
In patients with elevated conjugated bilirubin levels but otherwise normal liver function findings, the diagnosis of Dubin-Johnson syndrome can be confirmed by demonstrating an increase in the ratio of urinary coproporphyrin I to coproporphyrin III; type I makes up 80%, rather than the usual 25%, of the urinary coproporphyrin content in these patients.
Patients with Dubin-Johnson syndrome tend to have unique findings on hepatobiliary scintigraphy scans, demonstrating a combination of intense and prolonged visualization of the liver and delayed or failed visualization of the gallbladder.
See Workup for more detail.
Management
Dubin-Johnson syndrome is a benign disorder that requires no specific therapy, although patients should be warned that pregnancy, oral contraceptive use, administration of estrogens as a component of gender reassignment, and intercurrent illnesses can exacerbate the associated jaundice.
In the past, patients were treated with phenobarbital, which was used primarily to reduce serum bilirubin levels. This treatment is no longer recommended.
Rifampicin and ursodeoxycholic acid (UDCA) therapy have beneficial effects in chronic cholestatic diseases. These may result, in part, from the induction of MRP2 expression in the liver and kidney.
However, neither an indication nor a general role for these 2 agents has been defined in Dubin-Johnson syndrome. Rifampicin and UDCA should, in fact, be used with caution in patients with the disease, since they may actually increase conjugated bilirubinemia and bile acid levels in such cases. [2]
See Treatment and Medication for more detail.
Background
First described in 1954, [3] Dubin-Johnson syndrome (DJS) is an inherited, relapsing, benign disorder of bilirubin metabolism. This rare autosomal recessive condition is characterized by conjugated hyperbilirubinemia with normal liver transaminases, a unique pattern of urinary excretion of heme metabolites (coproporphyrins), and the deposition of a pigment that gives the liver a characteristic black color (see the image below). (See Presentation and Workup.)
Dubin-Johnson Syndrome. Gross liver specimen from a patient with Dubin-Johnson syndrome showing multiple areas of dark pigmentation. Image courtesy of Cirilo Sotelo-Avila, MD.
https://emedicine.medscape.com/article/173517-overview#a5
The primary defect in Dubin-Johnson syndrome is a mutation in an apical canalicular membrane protein responsible for the excretion of bilirubin and other nonbile salt organic anions. The protein was originally termed the canalicular multiple organic anion transporter (cMOAT) but is also known as multidrug resistance protein 2 (MRP2); it is a member of the ABC transporter superfamily. [4, 5, 6, 7, 8, 9, 10] (See Pathophysiology and Etiology.)
The clinical onset of Dubin-Johnson syndrome is most often seen in early adulthood; however, a neonatal onset has also been rarely described. Because of possible recurrence and second attacks of jaundice in later life, the neonatal form requires closer long-term follow-up. [11] (See Epidemiology.)
Hereditary hyperbilirubinemias can be divided into conjugated and unconjugated forms, and they may be caused by increased bilirubin production or decreased bilirubin clearance. [12] Examples are as follows [13] :
-
Conjugated hyperbilirubinemias: Dubin-Johnson syndrome and Rotor syndrome
-
Unconjugated hyperbilirubinemias: Gilbert syndrome and Crigler-Najjar syndrome
The conjugated and unconjugated hyperbilirubinemias are also classified as being, respectively, directly reacting and indirectly reacting. Directly reacting bilirubin reacts quickly with diazotized sulfanilic acid, forming a colored azodipyrrole, while indirectly reacting bilirubin reacts very slowly with the acid unless an accelerator, such as ethanol, is present.
Both inherited conjugated hyperbilirubinemias have a relatively benign course. However, diagnosing these conditions allows the physician to exclude more serious causes of hyperbilirubinemia and, thus, avoid unnecessary investigations and procedures. (See Presentation, Workup, and Treatment.)
Rarely, combined Dubin-Johnson and Gilbert syndromes can exist (“dual hereditary jaundice”), owing to compound anomalies in bilirubin conjugation and transport. [14] One study comprising 56 affected members across seven apparently unrelated Roma families revealed a novel deletion in the ABCC2 gene (homozygous novel variant c.1013_1014delTG in the eighth exon of ABCC2) in 17 individuals, as well as a homozygous dual defect (NG_011798.1:c.[1013_1014delTG]; NG_002601.2:g.[175492_175493insTA]) in 4 people, and a common 86 kbp haplotype encompassing promoter and part of the ABCC2 coding region among all families. [14]
Pathophysiology and Etiology
Dubin-Johnson syndrome (DJS) is an autosomal recessive disorder that is caused by a mutation in the ABCC2 gene, which comprises 32 exons, and is mainly expressed in the liver. [15] The gene encodes a 190-kD glycoprotein called human canalicular multispecific organic anion transporter (cMOAT) protein, also known as multidrug resistance protein 2 (MRP2) or ABCC2. [4, 5, 6, 7, 8, 9, 10, 15] This protein is mainly localized in the canalicular or apical membrane of hepatocytes and mediates adenosine triphosphate (ATP)-dependent transport of certain organic anions across the canalicular membrane of the hepatocyte, as well as exports conjugated bilirubin into bile. [9]
The cMOAT/MRP2/ABCC2 protein is encoded by a single-copy gene, MRP2/cMOAT/ABCC2, on chromosome 10q24. [16]
The conjugated hyperbilirubinemia observed in Dubin-Johnson syndrome results from defective transport of bilirubin glucuronide across the membrane that separates the hepatocyte from the bile canaliculi. Pigment that is not secreted from the hepatocyte is stored in the lysosome and causes the black liver color.
A hallmark of Dubin-Johnson syndrome, the mechanism of which is not fully understood, is a reversal of the usual ratio between the byproducts of heme biosynthesis: urinary coproporphyrin I levels are higher than coproporphyrin III levels. In unaffected individuals, the ratio of coproporphyrin III to coproporphyrin I is approximately 3-4:1. [17]
MRP2
MRP2 plays an important role in the detoxification of many drugs by transporting a wide range of compounds, especially conjugates of glutathione, glucuronate, and sulfate, which are collectively known as phase II products of biotransformation. Unlike other members of the MRP/ABCC family, MRP2 is expressed only on the apical membrane domain of polarized cells. Besides hepatocytes, MRP2 is located in renal proximal tubular cells, enterocytes, and syncytiotrophoblasts of the placenta. [18]
Energy derived from ATP is critical for the secretory function of MRP2. Mutations in the ATP-binding region of MRP2 represent a significant proportion of the recognized genetic defects in Dubin-Johnson syndrome. [19, 20]
The role of MRP2 in drug distribution, excretion, and interactions is not well-defined. There are no reports of drug toxicity specific to patients with Dubin-Johnson syndrome. However, in animal studies including Abcc2 (−/−) genetically engineered mice and in rat hepatocytes treated with MRP2- specific small interfering RNAs, enhanced toxicity and/or altered excretion was observed for various drugs, including erythromycin, human immunodeficiency virus protease inhibitors, and others. [21] Thus, caution should be employed when prescribing these agents in affected patients, and decreased dosing may be prudent.
Genetic mutations of ABCC2
According to the Human Gene Mutation Database (HGMD), there are 127 reported ABCC2 gene mutations. [22] Of those, 38 have been described in patients with Dubin-Johnson syndrome. [22] The nature of ABCC2 pathogenic variants is diverse, and it includes deletions, insertions, missense, nonsense, and splice junction mutations. [15, 21, 23, 24]
The majority of the Dubin-Johnson syndrome-causing mutations in ABCC2 are related to defects in MRP2 protein synthesis, localization or secretion activities. [24] Some mutations lead to rapid degradation of the mutant mRNA, whereas others affect protein maturation, protein stability, or the function of correctly localized proteins. [23, 24, 25]
Given the fact that numerous patients with Dubin-Johnson syndrome carry very rare variants in the homozygous state, consanguinity is known to play a crucial role in this rare autosomal recessive disease. [15] However, besides homozygous variants, in some populations there is a high proportion of affected individuals with clinical Dubin-Johnson syndrome who present as compound heterozygous mutations. [23] A report from China described that among the 30 patients with genetically confirmed diagnosis of Dubin-Johnson syndrome, 17 patients (57%) were homozygotes and 13 (43%) were compound heterozygotes for ABCC2 variants. [23]
The correlation between specific pathogenic variants of ABCC2 and phenotypes (age of onset, neonatal- vs adult-onset Dubin-Johnson syndrome) are only beginning to be defined. [21] Mutations in regions important for adenosine triphosphate (ATP) binding (the ATP-binding cassettes [ABC]) or mutations resulting in severely dysfunctional or absent protein (eg, splicing, truncating, and frameshift variants) are common in neonatal Dubin-Johnson syndrome, whereas variants involving domains other than the ABC of the MRP2 protein are commonly observed in adults. [26, 27]
Several studies have shown that variants vary across different ethnic groups. The p.Gly758Val variant is the most common one in Saudi patients, whereas the p.R768W variant occurs with a high frequency in the Japanese population. The p.Arg768Trp is the most common variant in the Korean population, and the p.R393W and p.Y1275X variants are known as disease-causing variants in Taiwan. Some variants appear to be unique to the Jewish population of Iranian and Moroccan origins (p.I1173F and p.R1150H, respectively). [27]
Epidemiology
Occurrence in the United States
The overall prevalence of Dubin-Johnson syndrome (DJS) is extremely low. However, although no accurate prevalence figures are available, it is known to be far more common than Rotor syndrome.
Race-, sex-, and age-related demographics
Dubin-Johnson syndrome has been described in all nationalities, ethnic backgrounds, and races. The highest recognized prevalence of the disease (1 case per 1300 population) is in Iranian Jews and is clustered in the same families. [28] This group may have an associated deficiency in clotting factor VII that is not observed in other populations. [29] The prevalence in Moroccan Jews is nearly as high, a reflection of the fact that these populations diverged about 2000-2500 years ago. [29]
Dubin-Johnson syndrome occurs in both sexes, but some authors have reported an increased incidence and earlier onset in males. [28]
This condition is rarely detected before puberty, although neonatal cases have been reported. It is most often diagnosed in the late teens and early adulthood.
Neonatal Dubin-Johnson syndrome
The clinical presentation of Dubin-Johnson syndrome as neonatal cholestasis is rare. [30] Typical patients with the neonatal-onset syndrome are likely to be full-term, well-looking neonates in whom cholestasis manifests in the first weeks of life, which then usually resolves within age 3–6 months, followed by a benign course. [27] Features such as failure to thrive, hepatomegaly, or splenomegaly are usually absent, and this disease generally does not affect the growth and development of children. [21, 31]
Affected neonates have increased serum total and direct bilirubin concentrations, along with normal liver synthetic function and normal to slightly elevated transaminases. [21, 27, 30, 32] Levels of aspartate transaminate (AST) and alanine aminotransferase (ALT) are significantly lower in infants with Dubin-Johnson syndrome than in those with neonatal cholestasis from other causes. [30, 32, 33, 34]
Total bilirubin levels in neonatal Dubin-Johnson syndrome are higher relative to levels in the adult syndrome. The two main reasons are reduced MRP2 expression during the neonatal period (1/200 of that in adults), as well as the decreased activity of UGT1A1 enzyme in newborns (< 1% of that in adults). [32]
As with adults, the urine coproporphyrin analysis is a reliable tool and the most helpful noninvasive test for the diagnosis of Dubin-Johnson syndrome in infants. [27, 30] A percentage of urinary coproporphyrin isomer I > 80% would be consistent with a diagnosis of Dubin-Johnson syndrome. [27]
Togawa et al demonstrated that only 38% (3) of the eight Dubin-Johnson syndrome patients who underwent liver biopsy had black livers or melanin-like pigment deposits in hepatocytes. [21] This age-related difference suggests that melanin-like pigment deposits in hepatocytes in patients with Dubin-Johnson syndrome may accumulate only gradually after birth. [21, 26] Therefore, unlike in the adult syndrome, it may be difficult to identify characteristic findings of Dubin-Johnson syndrome through liver biopsy in the neonatal condition. [33]
The neonatal form requires close long-term follow-up because of a possible biphasic appearance of cholestasis or recurrent episodes of jaundice. [21, 27] Failure to diagnose Dubin-Johnson syndrome early in the neonatal period may lead to diagnostic delay; some patients may be diagnosed in adulthood. Early diagnosis through a neonatal cholestasis gene panel can provide an accurate diagnosis of neonatal Dubin-Johnson syndrome and prevent unnecessary evaluation in the future. [15, 33]
Prognosis
Dubin-Johnson syndrome (DJS) is a benign condition, and life expectancy among patients is normal. The risk of liver cirrhosis and tumors is extremely low, and the prognosis is good. [32] An interesting case report describes an infant who received a living related liver transplant donor graft from his mother, who had Dubin-Johnson syndrome. One year after transplantation there were no unexpected issues with the donor or the child who had "inherited" Dubin-Johnson syndrome from his mother. [35]
Complications
Complications of Dubin-Johnson syndrome include jaundice (the most consistent finding) and hepatomegaly. Oral contraceptives, pregnancy, administration of estrogens as a component of gender reassignment, and intercurrent illness may exacerbate jaundice. Reduced prothrombin activity, resulting from lower levels of clotting factor VII, is found in 60% of patients.
Some neonates present with cholestasis, which may be severe. Increased fetal wastage was reported in a study. In a case report, cholecystolithiasis and choledocholithiasis developed in the presence of Dubin-Johnson syndrome. [36]
-
Dubin-Johnson Syndrome. Gross liver specimen from a patient with Dubin-Johnson syndrome showing multiple areas of dark pigmentation. Image courtesy of Cirilo Sotelo-Avila, MD.
-
Dubin-Johnson Syndrome. Microscopic histology of the liver in Dubin-Johnson syndrome showing multiple areas of granulated pigment. Fontana Mason stain. Image courtesy of Cirilo Sotelo-Avila, MD.
-
Dubin-Johnson Syndrome. Plain abdominal radiograph from a patient with a clinical diagnosis of acute cholecystitis. The diagnosis was confirmed with abdominal ultrasonography. The radiograph shows faint opacities in the region of the gallbladder fossa and dilated loops of small bowel in the epigastrium and the midabdomen secondary to localized ileus.
-
Dubin-Johnson Syndrome. A 26-year-old man known to be human immunodeficiency virus (HIV) positive presented with pain in the right upper quadrant and mild jaundice. This axial sonogram through the gallbladder (GB) and pancreas (P) shows sludge within the gallbladder and the lower common bile duct (CBD) (arrow). A diagnosis of acalculous cholecystitis was confirmed. A = aorta; IVC = inferior vena cava; S = splenic vein.




