Practice Essentials
DiGeorge syndrome (DGS) is one of a group of phenotypically similar disorders—including velocardiofacial syndrome (VCFS, or Shprintzen syndrome) and conotruncal anomaly face (CTAF) syndrome—that share a microdeletion of chromosome 22q11.2, a region known as the DGS critical region (see the image below). All these syndromes, because of their overlapping features, are now designated as a 22q11.2 deletion syndrome (22q11.2DS) and in the rest of the article will be referred to as 22q11.2DS.
Although the prognosis for 22q11.2DS varies widely, depending largely on the nature and degree of involvement of different organs, many adults live long and productive lives.
Signs and symptoms
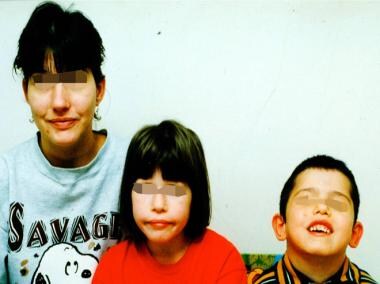
Patients with 22q11.2 DS usually have characteristic facial features. Common ones include the following (see the images below) [1] :
-
Retrognathia or micrognathia
-
Long face
-
High and broad nasal bridge
-
Narrow palpebral fissures
-
Small teeth
-
Asymmetrical crying face
-
Downturned mouth
-
Short philtrum
-
Low-set, malformed ears
-
Hypertelorism
-
Dimple on the tip of the nose
Congenital heart defects, a cleft palate or incompetence of the soft palate, and immune deficiencies are common. Patients may have short stature and occasional instances of growth hormone deficiency. Renal, pulmonary, gastrointestinal (GI), skeletal, and ophthalmologic abnormalities can also occur.
Children and adults with 22q11.2DS have high rates of behavioral, psychiatric, and communication disorders. In children, these include attention-deficit/hyperactivity disorder, anxiety, autism, and affective disorders. Adults have a high rate of psychotic disorders, particularly schizophrenia.
See Clinical Presentation for more detail.
Diagnosis
Genetic studies
-
Chromosomal microarray analysis (CMA) or array comparative genomic hybridization (aCGH)
-
Fluorescent in situ hybridization (FISH)
-
TBX1 gene studies
-
Multiplex ligation-dependent probe amplification (MLPA)
Additional laboratory tests
-
Complete blood cell (CBC) count
-
Serum calcium and parathyroid hormone (PTH) studies
Evaluation of T-cell count and function
-
Flow cytometry
-
Reverse-transcriptase polymerase chain reaction (RT PCR) assay to assess thymic T-cell out for detection of TCR excision circles (TREC)
-
Antibody response studies
Imaging studies
Imaging studies used in the diagnosis of thymic and cardiovascular abnormalities in 22q11.2DS include the following:
-
Radiography
-
Magnetic resonance imaging (MRI)
-
Computed tomography (CT) scanning
-
Echocardiography
-
Angiography and magnetic resonance angiography (MRA)
See Workup for more detail.
Management
Congenital heart defect
If a heart murmur and or other signs of a heart defect are present, consult a cardiologist right away, especially in the neonatal period.
Hypocalcemia
Begin calcium supplementation after proper tests (simultaneous serum calcium and serum PTH levels) are performed. Vitamin D supplementation may become necessary.
Immune reconstitution
Early thymus transplantation (ie, before the onset of infections) may promote successful immune reconstitution for subjects with complete absence of thymus (1% of 22q11.2DS subjects). A potential alternative treatment, adoptive transfer of mature T cells (ATMTC) through bone marrow transplantation has emerged as a successful therapy for 22q11.2DS.
For subjects with thymic hypoplasia, prophylactic antibiosis and antifungals are helpful for the first year of life. Management of autoimmune complications are important for older subjects.
Surgery
Cleft palate can be repaired with surgical modalities. [2]
Glottic web can be managed with surgical reconstruction or tracheotomy. [3]
Early intervention services
Monitor neurodevelopment and speech development, and refer the patient for educational therapies.
See Treatment and Medication for more detail.
Background
22q11.2DS (DiGeorge syndrome, or DGS) has a wide range of clinical features, including the following:
-
Abnormal facies
-
Congenital heart defects
-
Cognitive, behavioral, and psychiatric problems
-
Increased susceptibility to infections due to thymic aplasia or hypoplasia
Some collectively refer to these by the acronym CATCH-22 (cardiac defects, abnormal facies, thymic hypoplasia, cleft palate, and hypocalcemia resulting from 22q11.2 deletion). This designation has not been in use recently. See the image below. (See DDx and Workup.)
22q11.2DS encompasses the following phenotypically similar disorders—including DiGeorge syndrome:
-
Velocardiofacial syndrome (VCFS, or Shprintzen syndrome)
-
Cayler cardiofacial syndrome (asymmetric crying facies)
-
Conotruncal anomaly face (CTAF) syndrome
-
Some cases of autosomal dominant Opitz G/BBB syndrome
These syndromes were described as separate entities based on their prominent features and named as such prior to the discovery that they shared a common microdeletion of the DGS critical region, on chromosome 22 at band 22q11.2.
DiGeorge syndrome was originally described as a developmental field defect in the third and fourth branchial pouches, often presenting in the neonatal period with hypocalcemia and severe immune deficiency. Later, conotruncal heart defects were included. Velocardiofacial syndrome, on the other hand, was initially recognized as a syndrome of palatal defects, conotruncal heart defects, and characteristic facial features. (See Pathophysiology and Etiology.)
Partial versus complete 22q11.2DS based on immunologic features
Thymic hypoplasia or aplasia leading to defective T-cell function is one of the main features of 22q11.2DS. Depending on the T-cell proliferative responses to mitogens, the immunologic features of 22q11.2DS can be classified as partial or complete. Patients with partial 22q11.2DS have a below-normal proliferative response to mitogens, and the immune parameters may improve with time. Interleukin (IL)–7 may play a critical role in T-cell homeostasis in patients with partial 22q11.2DS. [4] However, in subjects with thymic hypoplasia, despite compensatory increase of T-cell numbers, TCR repertoire is reported to be decreased than normal controls.
Patients with complete 22q11.2DS are rare and have no T-cell responses to mitogens. These patients usually have very few detectable T cells in peripheral blood (1-2%) and usually require treatment of thymic transplant or hematopoietic stem cell transplantation. (See Treatment and Medication.)
B-cell defects
Although 22q11.2DS is categorized as a T-lymphocyte immunodeficiency, B-lymphocyte defects also occur. A review of 1023 patients with DGS revealed that 6% of patients older than 3 years had hypogammaglobulinemia and that 3% of patients with DGS were receiving immunoglobulin replacement therapy. [5]
Pathophysiology
The 22q11.2 deletion results in a range of embryonic developmental disruptions involving the head, neck, brain, skeleton, and kidneys. Portions of the heart, head and neck, thymus, and parathyroids derive from the third and fourth pharyngeal pouches, and this developmental field is disrupted due to the chromosomal microdeletion. This, in turn, leads to hypocalcemia, variable T-cell deficiency, and cardiac outflow defects. A combined T- and B-cell deficiency in part results from lack of T-helper cell function as typically seen in cases of complete 22q11.2DS.
Disease mechanism
The syndrome is caused by a microdeletion of band 22q11.2. The long arm of chromosome 22 (at q11) is prone to a microdeletion because of the presence of eight nonallelic, flanking, low-copy repeat DNA (deoxyribonucleic acid) sequence clusters (LCR22) labeled A–H. Clusters A–D are near the centromere. These repeat sequences lead to meiotic nonallelic crossing over between the 2 copies of chromosome 22 during spermatogenesis or oogenesis.
The most common deletion present in 85% of individuals is 3 million base pair (Mb) in size, extends from A to D, and encompasses approximately 40 genes and 4 micro RNAs. Among them is the TBX1 gene, suspected to play a major role in many of the typical features of this syndrome. There is some evidence that suggests that CNVs (copy number variants) and microRNAs in the rest of the genome likely influence the clinical variability seen even among the patients having the common deletion. [6] The remaining 15% of affected individuals have atypical smaller deletions including any of the LCR22 D–H.
Among other genes mapped in the deleted region that have been implicated in the pathogenesis of 22q11.2DS include HIRA (a transcriptional corepressor of cell cycle–dependent histone gene transcription and mammalian homologue of the yeast Hir1p and Hir2p proteins) and UFD1L (homologue of a highly conserved yeast gene involved in the degradation of ubiquitinated proteins).
Disease mechanism of immunodeficiency
The characteristic immunodeficiency in 22q11.2DS is a mild to moderate defect in T-cell lineage caused by thymic hypoplasia, typical of incomplete DGS. Naïve T-cell production is usually reduced with resultant low TREC (T-cell receptor excision circles) detected by PCR. Only a small fraction of patients present with marked impairment of T-cell function associated with a complete absence of thymus/T-cells (complete DGS), and severe systemic infections, consistent with severe combined immunodeficiency phenotype. Such patients can be detected by Newborn SCID Screening in the states where TREC enumeration is included in the newborn screening. Improvement with age in T-cell functions and numbers may be attributed to homeostatic T-cell proliferation secondary to limited T-cell production.
Variable secondary humoral defects, including hypogammaglobulinemia and selective antibody deficiency, may be present. This is attributed to impaired T-cell help, and su sequent impaired terminal B-cell maturation.
Disease mechanism of autoimmune diseases
Impaired T-cell production may predispose patients with 22q11.2 deletion to autoimmune diseases. In a cohort of 195 patients with 22q11.2DS, various autoimmune diseases, including juvenile rheumatoid arthritis, idiopathic thrombocytopenic purpura, and autoimmune hemolytic anemia, were more prevalent than in the age-matched general population. [7] No specific pattern of autoimmune disease appears to be associated with 22q11.2 deletion.
The frequency of autoimmune disorders in patients with partial 22q11.2DS was reviewed by Tison et al [8] in a large cohort of pediatric patients, and in that review, cytopenias and hypothyroidism were reported to be the most common autoimmune conditions. Autoimmunity was found in 10 (8.5%) of 130 patients, a frequency similar to that seen in a previous study in a different institution. Children with high or normal naive CD4 T-cell counts early in childhood had a lower risk of autoimmune disease.
Association with Graves disease has been reported sporadically. [9, 10] Other associated diseases include immune cytopenias, [11] immune thrombocytopenic purpura, [12] juvenile rheumatoid arthritis–like polyarthritis, [13] autoimmune uveitis, [14] and severe eczema. [15]
DiGeorge syndrome and velocardiofacial syndrome (VCFS) have also been found to be significantly associated with asthma but not with allergic rhinitis. [16]
A higher frequency of autoimmune diseases in 22q11.2DS patients is partly attributed to suppressed expression of AIRE (autoimmune regulator) in the thymic epithelial cell due to thymic hypolasia (low T-cell numbers), resulting in suppressed negative selection of autoreactive T cells in the thymus.
Disease mechanisms of neuropsychiatric disorders
The 22q11.2 microdeletion is the strong known genetic risk factor for schizophrenia and has been implicated with microRNA (miRNA)-mediated dysregulation. Two candidate genes for this condition are DiGeorge syndrome critical region gene 8 (DGCR8), which encodes a component of the microprocessor complex essential for miRNA biogenesis and miR-185. [17] miR-185 is reported to be down-regulated in brains of patients with idiopathic schizophrenia, and also is reported to be down-regulated in patients with 22q11.2 DiGeorge syndrome. [18]
Mode of inheritance
The occurrence of 22q11.2DS is sporadic in more than 90% of cases, being the result of de novo (noninherited) deletions. About 10% have inherited the deletion from a parent as an autosomal dominant condition. Sibling involvement has been observed only if a chromosome 22 deletion has been found in a parent. The hereditary cases show no predilection in inheritance from the mother or father, and an affected person has a 50% chance of transmitting the condition to his or her child. Wide intrafamily and interfamily variability in clinical manifestations is seen.
Epidemiology
Estimates of the incidence of 22q11.2DS range from 1 per 4000 to 1 per 7000 births. [19, 20] These estimates are based on a few population-based screening studies done in the 1990s and early 2000s and the diagnoses based on FISH technology. Thus, smaller deletions would have been missed. True prevalence can only be determined by uniform newborn screening.
Although 22q11.2DS is a congenital condition, the age at diagnosis is variable, being largely dependent on the severity and the types of associated birth defects. Thus, patients with more serious congenital cardiac defects or hypocalcemia are likely to be diagnosed in the neonatal period whereas those with only a submucous cleft palate and delayed speech, mild cardiac defects, normal immune function, or minimal facial anomalies are detected much later in childhood. Recurrent infections usually present in patients older than 3-6 months.
Late diagnosis into adulthood continues to be reported, especially in persons with isolated mild symptoms. Prenatal diagnosis in fetuses with a congenital heart anomaly has been made frequently and should be offered to a pregnant woman at risk of carrying a fetus with this syndrome.
Prognosis
The prognosis for 22q11.2DS varies widely, depending largely on the nature and degree of involvement of different organs, and it is important to note that many adults do live long and productive lives.
The most common cause of mortality in 22q11.2DS is a congenital heart defect and the second most common is severe immune deficiency. Mortality is higher in infancy because of the severity of these 2 conditions. Infants with thymus aplasia present with severe immunodeficiency and typically die of sepsis, caused by either bacterial or fungal infections.
In a large European collaborative study, 558 patients with 22q11.2DS were evaluated using a questionnaire. [21] Eight percent of the patients died, with more than half of the deaths occurring within the first month of life and the majority happening within 6 months of birth. Of the patients who survived, 62% had only mild learning problems or were developmentally normal. All the deaths except one were attributable to congenital heart disease. In this study, only 11% of patients were older than 18 years. Adult mortality data are limited. In one study, researchers compared survival of 102 adults (>17 yrs) with 22q11.2DS to survial of their 162 unaffected siblings. The study found survival in the affected group was reduced with an average age of death of 41.5 years (47.3 years in those without major congenital heart disease). [22]
Patient Education
Genetic counseling is essential to educate parents regarding the recurrence risk of 22q11.2DS. In addition, the families of patients with clinically significant immunodeficiency should be educated regarding the potential complications from exposure to live-attenuated vaccines that include rotavirus, MMR, and chicken pox vaccines.
Patients' families often feel alone after the syndrome is diagnosed. Because of its rarity, most parents have neither heard of this disorder nor do they know anyone who has it to whom they can turn to for support. Support groups and other resources are of invaluable help in this regard. Many written educational materials are available through various organizations, including those listed below.
International 22q11.2 Deletion Syndrome Foundation, Inc
PO Box 532
Matawan, NJ 07747USA
Telephone: 877-739-1849; email: info@22q.org
15 Meriden Ave
Stourbridge, West Midlands; DY8 4QN United Kingdom
Telephone: 0300-999-2211
National Library of Medicine Genetics Home Reference: 22q11.2 deletion syndrome
National Center for Biotechnology Information (NCBI) Genes and Disease: DiGeorge syndrome
Velo-Cardio-Facial Syndrome Education Foundation, Inc
PO Box 12591
Dallas, TX 75225, USA
Telephone 1-855-800-8237); email: info@vcfsef.org
108 Partinwood Drive
Fuquay-Varina, NC 27526, USA
Telephone: 919-567-8167; email: usinfo@c22c.org
-
Mother and children with 22q11.2 deletion syndrome.
-
An African American girl with 22q11.2 deletion syndrome.
-
The same child as in the previous image, showing an asymmetrical crying face.




