Practice Essentials
Congenital hypothyroidism is inadequate thyroid hormone production in newborn infants. It can occur because of an anatomic defect in the gland, an inborn error of thyroid metabolism, or iodine deficiency.
Signs and symptoms
Infants with congenital hypothyroidism are usually born at term or after term. Symptoms and signs include the following:
-
Decreased activity
-
Large anterior fontanelle
-
Poor feeding and weight gain
-
Small stature or poor growth
-
Jaundice
-
Decreased stooling or constipation
-
Hypotonia
-
Hoarse cry
Often, affected infants are described as "good babies" because they rarely cry and they sleep most of the time.
The physical findings of hypothyroidism may or may not be present at birth. Signs include the following:
-
Coarse facial features
-
Macroglossia
-
Large fontanelles
-
Umbilical hernia
-
Mottled, cool, and dry skin
-
Developmental delay
-
Pallor
-
Myxedema
-
Goiter
Anemia may occur, due to decreased oxygen carrying requirement. A small but significant number (3-7%) of infants with congenital hypothyroidism have other birth defects, mainly atrial and ventricular septal defects. [1]
See Presentation for more detail.
Diagnosis
Diagnosis of primary hypothyroidism is confirmed by demonstrating decreased levels of serum thyroid hormone (total or free T4) and elevated levels of thyroid-stimulating hormone (TSH). If maternal antibody–mediated hypothyroidism is suspected, maternal and neonatal antithyroid antibodies may confirm the diagnosis. [2] Such antibodies are an uncommon cause of congenital hypothyroidism. [3, 4]
The combination of low or low-normal serum total T4 levels and a serum TSH within the reference range suggests thyroid-binding globulin (TBG) deficiency. This congenital disorder causes no pathologic consequence but should be recognized to avoid unnecessary thyroid hormone administration.
Thyroid scanning
Thyroid scanning is not required to make or confirm the diagnosis of congenital hypothyroidism, but it can provide important information about the etiology.
On thyroid scanning (using technetium-99m or iodine-123), the absence of radionuclide uptake suggests sporadic athyreotic hypothyroidism but can also occur when uptake is blocked by excess iodide or thyroid receptor–blocking antibodies. If no uptake is found on isotope scanning, thyroid ultrasonography may demonstrate thyroid tissue. [5]
Thyroid scans can also demonstrate the presence of an ectopic thyroid, such as a lingual or sublingual gland, which is also sporadic. The presence of a bilobed thyroid in the appropriate position or a goiter would suggest either an inborn error of thyroid hormone production or transient hypothyroidism or hyperthyrotropinemia.
Other imaging studies
Ultrasonography may be a reasonable alternative or addition to scintigraphy but may fail to reveal some ectopic glands. [6]
A lateral radiograph of the knee may be obtained to look for the distal femoral epiphysis; this ossification center appears at about 36 weeks' gestation, and its absence in a term or postterm infant indicates prenatal effects of hypothyroidism. [7]
See Workup for more detail.
Management
The mainstay in the treatment of congenital hypothyroidism is early diagnosis and thyroid hormone replacement. Optimal care may include diagnosis before age 10-13 days and normalization of thyroid hormone blood levels by age 3 weeks. [8, 9]
Only levothyroxine is recommended for treatment. [10] Parents should be provided the hormone in pill form and taught proper administration, as follows:
-
The pills can be crushed in a spoon; dissolved with a small amount of breast milk, water, or other liquid immediately before administration; and administered to the child with a syringe or dropper
-
The pills should not be mixed in a full bottle of formula
Toddlers typically chew the tablets without problems or complaints.
Initial dosages of 10-15 mcg/kg/day, equivalent to a starting dose of 50 mcg in many newborns, have been recommended. [11] Equally good developmental results, but with higher thyroid-stimulating hormone (TSH) levels, have been reported with half this starting dose (25 mcg/day). [12]
See Treatment and Medication for more detail.
Background
Congenital hypothyroidism (CH) is inadequate thyroid hormone production in newborn infants. It can occur because of an anatomic defect in the gland, an inborn error of thyroid metabolism, or iodine deficiency. CH is the most common neonatal endocrine disorder; thyroid dysgenesis is thought to account for approximately 80-95%& of cases. [13] Some infants identified as having primary congenital hypothyroidism may have transient disease and not permanent congenital hypothyroidism. [5] See the image below.
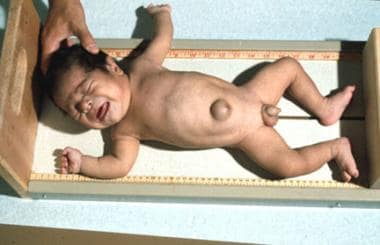 Congenital Hypothyroidism. An infant with cretinism. Note the hypotonic posture, coarse facial features, and umbilical hernia.
Congenital Hypothyroidism. An infant with cretinism. Note the hypotonic posture, coarse facial features, and umbilical hernia.
The term endemic cretinism is used to describe clusters of infants with goiter and hypothyroidism in a defined geographic area. Such areas were discovered to be low in iodine, and the cause of endemic cretinism was determined to be iodine deficiency. In the 1920s, adequate dietary intake of iodine was found to prevent endemic goiter and cretinism. [14] Endemic goiter and cretinism are still observed in some areas, such as regions of Bangladesh, Chad, China, Indonesia, Nepal, Peru, and Zaire.
The term sporadic cretinism was initially used to describe the random occurrence of cretinism in nonendemic areas. The cause of these abnormalities was identified as nonfunctioning or absent thyroid glands. This led to replacement of the descriptive term sporadic cretinism with the etiologic term congenital hypothyroidism. Treatment with thyroid replacement therapy was found to elicit some improvement in these infants (see the images below), although many remained impaired.
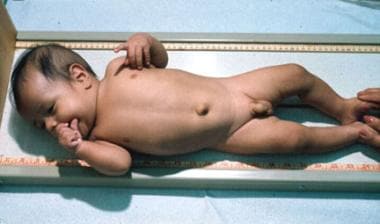 Congenital Hypothyroidism. An infant shown a few months after starting thyroid hormone replacement.
Congenital Hypothyroidism. An infant shown a few months after starting thyroid hormone replacement.
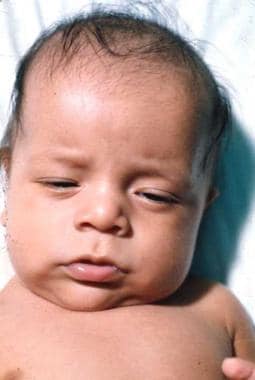 Congenital Hypothyroidism. Infant a few months after starting thyroid hormone replacement.
Congenital Hypothyroidism. Infant a few months after starting thyroid hormone replacement.
The morbidity from congenital hypothyroidism can be reduced to a minimum by early diagnosis and treatment. [15] Although initial preliminary studies were performed using thyroid-stimulating hormone (TSH) levels in cord blood, [16, 17] mass screening was made feasible by the development of radioimmunoassay for TSH and thyroxine (T4) from blood spots on filter paper, obtained for neonatal screening tests. [18, 19]
Pathophysiology
The thyroid gland develops from the buccopharyngeal cavity between 4 and 10 weeks' gestation. The thyroid arises from the fourth branchial pouches and ultimately ends up as a bilobed organ in the neck. Errors in the formation or migration of thyroid tissue can result in thyroid aplasia, dysplasia, or ectopy. By 10-11 weeks' gestation, the fetal thyroid is capable of producing thyroid hormone. By 18-20 weeks' gestation, blood levels of T4 have reached term levels. The fetal pituitary-thyroid axis is believed to function independently of the maternal pituitary-thyroid axis.
The thyroid gland uses tyrosine and iodine to manufacture T4 and triiodothyronine (T3). Iodide is taken into the thyroid follicular cells by an active transport system and then oxidized to iodine by thyroid peroxidase. Organification occurs when iodine is attached to tyrosine molecules attached to thyroglobulin, forming monoiodotyrosine (MIT) and diiodotyrosine (DIT). The coupling of two molecules of DIT forms tetraiodothyronine (ie, T4). The coupling of one molecule of MIT and one molecule of DIT forms T3. Thyroglobulin, with T4 and T3 attached, is stored in the follicular lumen. TSH activates the enzymes needed to cleave T4 and T3 from thyroglobulin. In most situations, T4 is the primary hormone produced by and released from the thyroid gland.
Inborn errors of thyroid metabolism can result in congenital hypothyroidism in children with anatomically normal thyroid glands.
T4 is the primary thyronine produced by the thyroid gland. Only 10-40% of circulating T3 is released from the thyroid gland. The remainder is produced by monodeiodination of T4 in peripheral tissues. T3 is the primary mediator of the biologic effects of thyroid hormone and does so by interacting with a specific nuclear receptor. Receptor abnormalities can result in thyroid hormone resistance.
The major carrier proteins for circulating thyroid hormones are thyroid-binding globulin (TBG), thyroid-binding prealbumin (TBPA), and albumin. Unbound, or free, T4 accounts for only about 0.03% of circulating T4 and is the portion that is metabolically active. Infants born with low levels of TBG, as in congenital TBG deficiency, have low total T4 levels but are physiologically normal. Familial congenital TBG deficiency can occur as an X-linked recessive or autosomal recessive condition.
The contributions of maternal thyroid hormone levels to the fetus are thought to be minimal, but maternal thyroid disease can have a substantial influence on fetal and neonatal thyroid function. Immunoglobulin G (IgG) autoantibodies, as observed in autoimmune thyroiditis, can cross the placenta and inhibit thyroid function. Thioamides used to treat maternal hyperthyroidism can also block fetal thyroid hormone synthesis. Most of these effects are transient. Radioactive iodine administered to a pregnant woman can ablate the fetus's thyroid gland permanently.
The importance of thyroid hormone to brain growth and development is demonstrated by comparing treated and untreated children with congenital hypothyroidism. Thyroid hormone is necessary for normal brain growth and myelination and for normal neuronal connections. The most critical period for the effect of thyroid hormone on brain development is the first few months of life. [15]
Etiology
Endemic cretinism is caused by iodine deficiency and is occasionally exacerbated by naturally occurring goitrogens. [20] Dysgenesis of the thyroid gland, including agenesis (ie, complete absence of thyroid gland) and ectopy (lingual or sublingual thyroid gland), may be a cause.
Inborn errors of thyroid hormone metabolism include dyshormonogenesis. Most cases are familial and inherited as autosomal recessive conditions. These may also include the following:
-
Thyroid-stimulating hormone (TSH) unresponsiveness (ie, TSH receptor abnormalities) [21]
-
Impaired ability to uptake iodide
-
Peroxidase, or organification, defect (ie, inability to convert iodide to iodine)
-
Pendred syndrome, a familial organification defect associated with congenital deafness
-
Thyroglobulin defect (ie, inability to form or degrade thyroglobulin)
-
Deiodinase defect
Thyroid hormone resistance (ie, thyroid hormone receptor abnormalities) may also be a cause. [21]
In maternal autoimmune disease, transplacental passage of antibodies cause transient or permanent hypothyroidism. [2, 22]
Radioactive iodine therapy of pregnant women may cause permanent congenital hypothyroidism. Iodine in contrast agents or skin disinfectants can cause hypothyroidism or hyperthyrotropinemia in premature neonates. [23]
TSH or thyrotropin-releasing hormone (TRH) deficiencies are also noted. Hypothyroidism can also occur in TSH or TRH deficiencies, either as an isolated problem or in conjunction with other pituitary deficiencies (eg, hypopituitarism). If present with these deficiencies, hypothyroidism is usually milder and is not associated with the significant neurologic morbidity observed in primary hypothyroidism.
Although the etiology of congenital hypothyroidism with thyroid gland-in-situ (GIS) is not completely understood, mutations in eight known causative genes (TG, TPO, DUOX2, DUOXA2, SLC5A5, SLC26A4, IYD, and TSHR) have been identified. [24, 25]
Epidemiology
Studies have reported a change in the epidemiology, with a doubling in incidence to around 1 in 1500 live newborns. [13, 26] This is thought to be due in part to an increase in congenital hypothyroidism with thyroid gland-in-situ (GIS). [24] Lower TSH screening cutoffs may also be driving this increase in diagnosis, although altered ethnicities of the screened population, increased multiple and premature births, and iodine status are contributing factors. [27]
Congenital hypothyroidism is more common in infants with birthweights less than 2,000 g or more than 4,500 g. [28, 29]
An increased incidence of congenital hypothyroidism is observed in twins. [30, 28, 31] Twin births are approximately 12 times as likely to have congenital hypothyroidism as singletons. [32] Usually, only one twin is hypothyroid, but a common in-utero exposure can cause hypothyroidism in both. [33]
Most studies of congenital hypothyroidism suggest a female-to-male ratio of a 2:1. Devos et al showed that much of the discrepancy is accounted for by infants with thyroid ectopy. [34]
In central Africa, where iodine deficiency occurs along with excess dietary cyanate from cassava (Manihot esculenta), [35] as many as 10% of newborns may have both low cord blood T4 concentration and TSH concentrations over 100 mU/L. [36]
Some of the highest incidences have been reported from various locations in the Middle East. [37, 38, 39, 40]
Prognosis
Congenital hypothyroidism does not affect the all-cause standardized mortality ratio in treated patients. [41] Profound intellectual disability is the most serious effect of untreated congenital hypothyroidism. Severe impairment of linear growth and bone maturation also occurs. Affected infants whose treatment is delayed can have neurologic problems such as spasticity and gait abnormalities, dysarthria or mutism, and autistic behavior.
Early diagnosis and treatment of congenital hypothyroidism prevents severe intellectual disability and other neurologic complications. [42] Even with early treatment, some children demonstrate mild delays in areas such as reading comprehension and arithmetic in third grade. Some of these delays improve by sixth grade. Despite treatment, individuals diagnosed by newborn screening as a group do not do as well as their euthyroid peers. [43]
Infants with delayed diagnosis or a longer time to normalize thyroid hormone levels have poorer outcomes. Although continued improvement in IQ has been documented in treated patients through adolescence, some cognitive problems may persist. These may include problems in visuospatial, language, and fine motor function. Defects in memory and attention may also be present.
Early studies of outcome suggested that infants without a distal femoral epiphysis did less well than those with one, although both groups had results in the normal range. [44] The author of this study was later unable to demonstrate an effect of bone age at diagnosis on outcome. [45] Another study was unable to demonstrate any difference in outcome in infants with or without a distal femoral epiphysis. [46]
Patient Education
Parents should be educated regarding their child's disorder, the potential problems associated with no treatment or inadequate treatment, and the benefits of early and appropriate treatment. This should include instructions on the proper administration of the medication and how and when to follow up with the physician. Because learning problems are possible, even with early diagnosis and treatment, parents should be advised when to seek psychomotor and educational evaluations and interventions. Early childhood intervention programs, if available, should be encouraged.
When inborn errors of thyroid hormone production are suspected, genetic counseling should be provided.
-
Congenital Hypothyroidism. An infant with cretinism. Note the hypotonic posture, coarse facial features, and umbilical hernia.
-
Congenital Hypothyroidism. Note the macroglossia.
-
Congenital Hypothyroidism. An infant shown a few months after starting thyroid hormone replacement.
-
Congenital Hypothyroidism. Infant a few months after starting thyroid hormone replacement.








