Background
The complement system exerts many of its effects through complement receptors (CRs). Of the 8 plasma membrane receptors for complement, only deficiencies of CR3 and CR4 due to CD18 deficiency have been described, known as leukocyte adhesion deficiency (LAD) type 1.
Table 1. Complement Receptors (Open Table in a new window)
Receptor |
Cluster Designation |
Ligand |
Cell Distribution |
Activity |
CR1 |
CD35 |
C3b/C4b |
RBC, polymorphonuclear cell, macrophage, B cell, follicular dendritic cell |
Immune adherence, phagocytosis |
CR2 |
CD21 |
C3dg/C3d |
B cell, follicular dendritic cell |
Co-receptor for B-cell signaling |
CR3 |
CD11b/CD18 |
C3bi, ICAM |
Myeloid |
Phagocytosis, immune adherence |
CR4 |
CD11c/CD18 |
C3bi, ICAM |
Myeloid |
Phagocytosis, immune adherence |
C1qRP |
None |
C1q, MBL, surfactant |
Polymorphonuclear cell, macrophage |
Promotes phagocytosis |
C3aR |
None |
C3a, C4a |
Polymorphonuclear cell, macrophage, epithelial cell, smooth-muscle cell |
Anaphylatoxin |
C4aR |
None |
C4a |
Polymorphonuclear cell, macrophage, epithelial cell, smooth-muscle cell |
Anaphylatoxin |
C5aR |
CD88 |
C5a |
Polymorphonuclear cell, macrophage, epithelial cell, smooth-muscle cell |
Anaphylatoxin |
ICAM = intercellular adhesion molecule, MBL = mannose-binding lectin |
||||
Diseases Related to the Complement System
CR1 (CD35)
Upon activation and cleavage of C3, C3b is formed as a major fragment that covalently binds to its target (see Table 1). C3b and C4b bind to CR1, which is present on various phagocytes and also on erythrocytes and B cells. CR1 participates in immune adherence and phagocytosis. [1] Immune adherence refers to the process by which bacteria coated with immunoglobulin G (IgG) or immunoglobulin M (IgM) antibody and C3b adhere to erythrocytes, which facilitates phagocytosis by neutrophils. No complete congenital deficiency of CR1 has been reported. Acquired forms of CR1 deficiency have been associated with autoimmune disorders, such as systemic lupus erythematosus, hemodialysis in patients with diabetic nephropathy, and preeclampsia. [2] CR1 deficiency may partly account for the increased likelihood of infection reported in thesepatients.Recombinanterythropoietin(rEPO)hasbeenreportedtoincreaseerythrocyteCR1levels.
CR2 (CD21)
CR2 binds C3dg, and C3d is present on B cells and dendritic cells (see the Table 1). CR2 associates with CD19 forming a CR2-CD19 complex when stimulated by C3d-bearing antigen engaging CR2. [3] Thus, it enhances and prolongs antigen signaling on B cells.
Mutation of CR2 (CD21) has been associated with common variable immunodeficiency (CVID) characterized by onset of infections in early childhood. Serum IgG levels are decreased but serum IgA and IgM levels are normal; antibody responses to polysaccharide antigens are decreased and antibody responses to protein antigens are slightly reduced. B cells are normal but memory and switch B cells are low. Treatment is with intravenous or subcutaneous gammaglobulin treatment. [4]
C1q receptor for phagocytosis (C1qRP)
Evidence suggests that C1q binds a receptor present on phagocytic cells, termed C1qRP. [5] C1q is a member of the collectin family, which also includes surfactant A, surfactant D, and mannose-binding lectin (MBL). See Table 1. C1qRP binds to MBL and surfactants. Surfactants and MBL play an important role in innate immunity. MBL deficiency manifests as increased susceptibility to polysaccharide-encapsulated bacteria, with subsequent recurrent respiratory tract infections, abscesses, sepsis, and meningitis. C1qRP deficiency has not been described.
C3a, C4a, and C5a (CD88) receptors
Receptors for C3a and C5a have been identified; whether a distinct receptor for C4a is present is unclear (see Table 1). The C3a receptor binds C3a and C4a. These receptors are present on phagocytic cells, mast cells, and lung epithelial and smooth muscle cells. [6] These receptors play a role in C3a-mediated and C5a-mediated anaphylactic reactions. [7] Deficiencies of these receptors have not been described.
CR3 (CD11b/CD18) and CR4 (CD11c/CD18)
The CD11/CD18 complex is part of the beta-2 integrin family and is important in adhesion and phagocytosis (see Table 1). [8] Deficiency of CD18 on phagocytic cells causes LAD type 1 (see Table 2). Three CD11 alpha chains and a common CD18 beta chain form heterodimer transmembrane complexes (CD11a/CD18, CD11b/CD18, CD11c/CD18). See Table 3 below. CD11a/CD18 is also known as leukocyte factor antigen-1 (LFA-1), CD11b/CD18 is known as CR3, and CD11c/CD18 is known as CR4.
Ligands for CD11a/CD18 are intercellular adhesion molecules (ICAMs), ligands for CD11b/CD18 are complement C3bi and ICAMs, and ligands for CD11c/CD18 are C3bi and ICAMs. CD18 deficiency results in loss of expression of LFA-1, CR3 (CD11b/CD18), and CR4 (CD11c/CD18) (see Table 3). These defects lead to abnormal neutrophil, macrophage, and T-cell and B-cell adhesion to vascular endothelium and subsequent migration into infectious sites. In addition, T- and B-cell functions are severely decreased.
Table 2. Leukocyte Adhesion Defects (Open Table in a new window)
Disease |
Inheritance |
Genetic Defect |
Protein Defect |
Affected Cells |
Affected Function |
Manifestations |
LAD type 1 |
Autosomal recessive |
INTGB2 |
CD18 |
Polymorphonuclear cell, macrophage, lymphocytes, NK cells |
Tight adherence, chemotaxis, endocytosis, T-cell/NK-cell cytotoxicity |
Delayed cord separation, skin ulcers, periodontitis, leukocytosis, poor pus formation |
LAD type 2 |
Autosomal recessive |
FUCT1 encoding for GDP-fucose transporter |
Fucosylated proteins, sialyl-Lewis X (sLeX, CD15s) |
Polymorphonuclear cell, macrophage |
Rolling, chemotaxis, tethering |
Same as LAD type 1 plus hh-blood group, intellectual disability |
LAD type 3 |
Autosomal recessive |
Kindlin 3 (FERMT3), involved in activation of integrin |
Kindlin 3 |
Polymorphonuclear cell, macrophage, lymphocytes, NK cells |
Tight adherence |
Same as LAD type 1 plus bleeding tendency |
Rac 2 deficiency |
Possibly autosomal dominant |
RAC2 |
Rac2, involved in regulation of actin cytoskeleton |
Polymorphonuclear cell, decreased TRECs |
Chemotaxis, O2- production |
Recurrent infections, poor wound healing, leukocytosis, poor pus formation |
E-selectin |
Possibly autosomal recessive |
Unknown |
E-selectin |
Endothelial cells |
Rolling, tethering |
Recurrent infections, poor pus formation, mild neutropenia |
NK = Natural killer, TRECs = T-cell receptor excision circles |
||||||
Table 3. Adhesion Molecules (Open Table in a new window)
Molecule |
CD Number |
Distribution |
Ligand |
Function |
Integrins |
||||
LFA-1 |
CD11a/CD18 |
All leukocytes |
ICAM-1, 2, 3 |
Adhesion, migration |
CR3 |
CD11b/CD18 |
Polymorphonuclear cell, macrophage, NK cells, eosinophils |
ICAM-1,2; C3bi |
Adhesion, migration |
CR4 |
CD11c/CD18 |
All leukocytes |
C3bi, ICAM-1, CD23, fibrinogen |
Adhesion |
Alpha4-beta7 |
None |
Lymphocytes, NK cells, eosinophils |
MadCAM-1, VCAM-1, fibronectin |
Adhesion, migration, rolling |
VLA-4 |
CD49d/CD29 |
Lymphocytes, NK cells, eosinophils, basophils |
VCAM-1, fibronectin |
Adhesion, migration, rolling |
Selectins |
||||
E |
CD62E |
Endothelial cells, platelets |
Sialylated, fucosylated molecules (sLeX, CD15s) expressed on PSGL-1 and ESL-1 |
Rolling |
P |
CD62P |
Endothelial cells, platelets |
Sialylated, fucosylated molecules (sLeX) expressed on PSGL-1 |
No data |
L |
CD62L |
Leukocytes |
Sialylated, fucosylated molecules (often sulfated) expressed on CD34, MadCAM-1 and other glycoproteins-1 |
Rolling |
MadCAM = Mucosal addressin cell adhesion molecule; VCAM = Vascular cell adhesion molecule; VLA = Very late activation antigen |
||||
Additional Leukocyte Adhesion Deficiency Syndromes
As seen in the image below, additional LAD syndromes have been identified that also interfere with the phagocytic cell adhesion cascade (see Table 2). [8]
LAD type 2 is due to decreased expression of fucosylated proteins, such as sialyl Lewis X (sLeX, CD15s), that are ligands for selectins necessary for the initiation of phagocytic cells attaching to endothelial cells in a process called rolling. LAD type 3 is due to defect of activation of Rap-1 important in the activation of integrins; this also results in defects of tight adherence. Rac2 deficiency results in decreased chemotaxis, superoxide anion production, and phagocytosis. E-selectin deficiency, a ligand for sialylated and fucosylated molecules, on endothelial cells results in decreased neutrophil rolling and tethering
LAD type 1, type 2, and type 3 are autosomal recessive disorders of neutrophils characterized by neutrophilia, recurrent severe bacterial infections, absence of inflammatory infiltrates, delayed umbilical-cord separation, and impaired wound healing (see Table 2). The defect in LAD type 1 is absent or defective expression of CD11/CD18 on the surface of neutrophils, macrophages, and lymphocytes.
Leukocyte adhesion deficiency type 1
Patients with LAD type 1 present either with a severe form with absence of CD18 or with a moderate form with 5–30% expression of CD18. [9, 10, 8] See Table 4. In the severe form, recurrent bacterial infections, skin infections, periodontitis, and gingivitis begin in the first year of life. Infections with Staphylococcus, Pseudomonas, Klebsiella, Enterococcus, and Proteus species and with Escherichia coli are common. Infectious sites are typically devoid of inflammatory cells because of the adhesion defect. Without immune reconstitution, death usually ensues when patients are younger than 2 years. In the moderate phenotype, the clinical course is much milder.
Table 4. Subtypes of Leukocyte Adhesion Deficiency Type 1 (Open Table in a new window)
Subtype |
mRNA level |
CD18 Expression |
Clinical Presentation |
1 |
None |
None |
Severe |
2 |
Low |
Trace |
Moderate |
3 |
Reference range |
Trace, small protein precursor |
Moderate |
4 |
Reference range |
Large protein precursor |
Severe |
5 |
Reference range |
Normal protein precursor |
Moderate |
mRNA = messenger RNA. |
|||
Leukocyte adhesion deficiency type 2
LAD type 2 deficiency is caused by defective fucosylation that leads to immunodeficiency and psychomotor retardation. [11, 12, 13, 14, 8] LAD type 2 is due to a defect in fucose metabolism that leads to deficiency of ligands for endothelial selectins, such as sLeX (CD15s), but normal expression of CD11/CD18 complexes (see Table 2). The defect leads to abnormal neutrophil rolling, although neutrophil adherence is normal (see Table 3). Also, T- and B-cell functions are normal.
LAD type 2 has been reported in approximately 4 families of Arabic origin. The clinical course is milder, characterized by severe periodontitis; however, severe infections are not usually observed (see Table 2). Other features of LAD type 2 include severe intellectual disability, distinctive facies, and short stature. The facial features include a broad and depressed nasal bridge, long eyelashes, and a simian crease, and dorsally positioned second toes are present. In addition, neither the H blood group antigen (Bombay phenotype) nor the Lewis blood type antigens (Lea and Leb) are expressed.
The genetic defect is due to defective Golgi-GDP-fucose transporter (GFTP). GFTP serves to transport the nucleotide sugar GDP-fucose into the Golgi lumen, where the sugar serves as a substrate for fucosylation reactions mediated by several fucosyl transferases. GFTP is a 364 aa protein with 10 transmembrane domains with carboxy and amino termini exposed to the cytosol. The defect leads to decreased fucosylated carbohydrate molecules, such as leukocyte sialyl Lewisx (sLex), which severely decreases interactions with endothelial selectins. This reduces selectin-mediated leukocyte tethering and rolling.
The reason hypofucosylation leads to abnormal neurodevelopment is unknown. One speculation is that the cause is decreased signaling through Notch, which is required for a number of development processes. Four Notch proteins (Notch 1-4) have been reported. They are cell-surface molecules that are cleaved after binding to ligands (Delta 1, 3, and 4 and Jagged 1, and 2). Notch receptors are O-fucosylated and critical for Notch-ligand binding. The cleaved Notch is then translocated into the nucleus, where it activates several genes implicated in developmental processes. L-fucose supplementation is the recommended treatment for patients with LAD type 2 and does improve immunodeficiency.
Leukocyte adhesion deficiency type 3
LAD type 3 was described in 2 Arab brothers and 18 Turkish patients with profound leukocytosis, recurrent infections with absence of pus, and platelet aggregation defects resulting in bleeding (see Table 2). [15] The neutrophils demonstrated normal neutrophil rolling and opsonophagocytosis but abnormal neutrophil chemotaxis and tight adherence, similar to that found in LAD type 1 deficiency.
The genetic defect was identified as stop codon mutations in Kindlin3, also known as FERMT3. [15] Kindlin3 is expressed exclusively in hematopoietic cells. Kindlin3 plays an essential role in the proper conformation of integrins to active form. Integrin activation starts by chemokine induction through membrane bound G-protein coupled receptors (GPCR) and transduction by CalDAG GEF1 and Rap1 intracellularly. Rap1-GTP-interacting adapter molecule (RIAM) then binds activated Rap1 to Talin which leads to binding to the cytoplasmic tail of the β-integrin chain. Kindlin3 enables Talin to induce a modification of integrins from an inactive bent conformation to an active open conformation.
Leukocyte adhesion deficiency type 1/variant syndrome
LAD type 1/variant syndrome consists of a moderate LAD type 1–like syndrome and a severe Glanzmannlike bleeding disorder. [8, 16] Thus, it clinically resembles LAD type 3. LAD type 1/variant syndrome is rare and only a few patients, predominantly of Turkish descent, have been described. The clinical picture consists of delayed cord detachment; recurrent bacterial, fungal, and cytomegalovirus infection, beginning early in infancy; and poor wound healing. Bleeding tendency is moderate to severe, requiring repeated platelet transfusions. Neutrophilia is not as severe as seen in LAD type 1, with a WBC count of 10,000-30,000 with 60-90% neutrophils.
Mutations of FERMT3 have been found to be caused by truncation mutations of FERMT3 (Kindlin3). [17] Neutrophil adhesion, chemotaxis, and zymosan-induced nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity are decreased. CD18 gene and protein expression are normal; Rap1, Rap2, and Rap regulatory activity are normal. GPCR-induced integrin activation is absent, similar to that seen in LAD type 3. Successful bone marrow transplantation has been performed in patients with LAD type 1/variant syndrome.
Rac2 deficiency
A male patient with a mutation of RAC2 was reported to be a cause of LAD. [18, 19] RAC2 is a member of the Rho family of guanosine triphosphatases (GTPases) critical in the regulation of the actin cytoskeleton and superoxide production. Upon clinical evaluation, the patient had profound leukocytosis, perirectal abscesses and poor wound healing with an absence of pus. Chemotaxis, superoxide anion production, phagocytosis, and neutrophil primary granule release were impaired. Accetta et al described a patient with markedly decreased TRECs identified on newborn screening who was subsequently diagnosed with Rac2 LAD. [20] Bone marrow transplantation was performed resulting in clinical cure and correction of neutrophil defects.
E-selectin deficiency
Another molecular defect causing LAD was described in a female patient with defective endothelial expression of E-selectin. [21] She had Pseudomonas omphalitis, recurrent ear and urinary tract infections, and severe soft tissue infections with poor pus formation. She also had mild neutropenia, but the number of neutrophils increased in response to infection and infusions of granulocyte-macrophage colony-stimulating factor (GM-CSF). Because of decreased E-selectin expression, defective rolling and tethering of phagocytic cells was suggested. [8]
Pathophysiology
The basis of LAD type 1 is various mutations in the common beta chain (CD18) of the beta-2 integrin family located on chromosome 21. [8, 22] Genes for the 3 CD11 chains (CD11a, CD11b, CD11c) are clustered on chromosome arm 16q. Defects in the beta chain result in the absence, insufficient amount, or abnormal function of the common CD18 unit. Two CD11 and 2 CD18 genes form the CD11/CD18 heterodimer complex. CD11/CD18 are members of the liver-cell adhesion molecule (LCAM) family, and their ligands are ICAMs and fibrinogen (see Table 3).
CD11a/CD18 is present on all leukocytes; CD11b/CD18 and CD11c/CD18 are present on neutrophils, macrophages, NK cells, and subsets of T cells and B cells (see Table 3). With these receptor-ligand interactions, these molecules play a crucial role in tight adhesion to endothelial vessel walls. In the initial adhesion step under conditions of blood flow, leukocytes begin a process of rolling. This is largely mediated by selectins, CD62E, CD62P, and CD62L, present on endothelial cells (see Table 3). Sialyl-Lewis X (sLeX, CD15s) is one of the counterligands. A defect in fucosylated proteins (eg, sLeX) that are ligands for selectins causes LAD type 2 and abnormal neutrophil rolling (see the image below). Absence of the neutrophil receptor for E-selectin (CD62E) results in a similar inability for neutrophils to migrate to inflammatory sites and respond to infections.
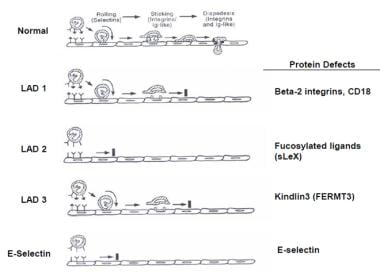 Protein defects.
Protein defects.
In the next step, neutrophils firmly adhere to the endothelial vessel wall and then transmigrate (see the image above). CD18 defects cause a marked decrease in firm neutrophil adherence. In addition, transmigration of neutrophils is abnormal. As a result, in infectious sites, inflammatory cells are scarce. Also, CD11/CD18 is involved with T-cell and B-cell and macrophage interactions; therefore, CD18 defects lead to decreased T-cell function and decreased CD8, NK, and antibody-dependent cell-mediated cytotoxicity (ADCC).
The diversity of the gene defects causes 5 subtypes of LAD type 1 in which genotype produces different phenotype expression (see Table 4):
-
Type 1 LAD1 produces no beta subunit mRNA, produces no CD18, and produces severe clinical disease.
-
Type 2 LAD1 has low levels of mRNA, trace CD18, and moderate clinical disease.
-
Type 3 LAD1 has reference range levels of mRNA and a small protein precursor, and it produces moderate clinical disease. (Types 2 and 3 have approximately 3-10% expression of CD11/CD18.)
-
Type 4 LAD1 has reference range levels of mRNA and a large protein precursor, and it produces severe clinical disease.
-
Type 5 LAD1 has reference range mRNA levels and a normal protein precursor, and it produces moderate disease.
Heterozygotes have approximately one half of the normal amounts of CD11/CD18 on phagocytic cells and lymphocytes and have no clinical disease.
Epidemiology
Frequency
Leukocyte adhesion deficiency type 1 is estimated to occur in 1 per million people worldwide. At least 300 cases of this condition have been reported in the scientific literature. [23]
Demographics
LAD syndromes affect males and females in equal numbers. The exact incidence of these disorders in the general population is unknown. LAD I is by far the more common one with several hundreds of patients reported in the medical literature from all over the world. LAD II is very rare reported in less than 10 patients and LAD III is also rare with 25 patients mainly from the Middle East region. These disorders often go unrecognized and may be misdiagnosed, making it difficult to determine their true frequency in the general population. LAD I was first described in the medical literature in 1979. LAD II was first reported in 1992. LAD III was first reported in 1997. [24]
The first clue to LAD type 1 may be the delayed separation of the umbilical cord. This is not manifested in LAD type 2. Patients with LAD type 1 with the severe phenotype are susceptible to infections beginning at birth and these infections typically occur by age 3–6 months. In LAD type 1 moderate phenotypes, infections are milder and may occur later.
Mortality/Morbidity
Five subtypes of LAD type 1 are recognized, with moderate-to-severe clinical phenotypes. [8] The severe phenotype of LAD type 1 is a life-threatening primary immunodeficiency with severe infections. A recent study noted that the incidence of primary immunodeficiencies markedly increased from 1976-2006. [25] Children rarely live past age 2 years without immune reconstitution. In the more mild-to-moderate phenotypes of LAD type 1, the clinical course is milder and the patients have a better prognosis. In both forms, wound healing is abnormal. Skin ulcers and/or necrotic lesions may form; skin grafts may be necessary. Abnormal dentition, with loss of deciduous and secondary teeth, occurs in all phenotypes of LAD.
LAD type 2 is associated with marked periodontitis. [8] Some patients with LAD type 2 have also had severe bacterial infections, similar to patients with LAD type1. In addition, patients with LAD type 2 often have short stature, delayed development, and intellectual disability.
Prognosis
The prognosis of patients with the LAD type 1 severe phenotype is poor; these patients require immune reconstitution.
Patients with mild and moderate phenotypes of LAD type 1 have infections of decreased severity.
CD18 expression of 10% seems to confer protection against invasive life-threatening infections. [9]
-
Protein defects.
Tables
Receptor |
Cluster Designation |
Ligand |
Cell Distribution |
Activity |
CR1 |
CD35 |
C3b/C4b |
RBC, polymorphonuclear cell, macrophage, B cell, follicular dendritic cell |
Immune adherence, phagocytosis |
CR2 |
CD21 |
C3dg/C3d |
B cell, follicular dendritic cell |
Co-receptor for B-cell signaling |
CR3 |
CD11b/CD18 |
C3bi, ICAM |
Myeloid |
Phagocytosis, immune adherence |
CR4 |
CD11c/CD18 |
C3bi, ICAM |
Myeloid |
Phagocytosis, immune adherence |
C1qRP |
None |
C1q, MBL, surfactant |
Polymorphonuclear cell, macrophage |
Promotes phagocytosis |
C3aR |
None |
C3a, C4a |
Polymorphonuclear cell, macrophage, epithelial cell, smooth-muscle cell |
Anaphylatoxin |
C4aR |
None |
C4a |
Polymorphonuclear cell, macrophage, epithelial cell, smooth-muscle cell |
Anaphylatoxin |
C5aR |
CD88 |
C5a |
Polymorphonuclear cell, macrophage, epithelial cell, smooth-muscle cell |
Anaphylatoxin |
ICAM = intercellular adhesion molecule, MBL = mannose-binding lectin |
||||
Disease |
Inheritance |
Genetic Defect |
Protein Defect |
Affected Cells |
Affected Function |
Manifestations |
LAD type 1 |
Autosomal recessive |
INTGB2 |
CD18 |
Polymorphonuclear cell, macrophage, lymphocytes, NK cells |
Tight adherence, chemotaxis, endocytosis, T-cell/NK-cell cytotoxicity |
Delayed cord separation, skin ulcers, periodontitis, leukocytosis, poor pus formation |
LAD type 2 |
Autosomal recessive |
FUCT1 encoding for GDP-fucose transporter |
Fucosylated proteins, sialyl-Lewis X (sLeX, CD15s) |
Polymorphonuclear cell, macrophage |
Rolling, chemotaxis, tethering |
Same as LAD type 1 plus hh-blood group, intellectual disability |
LAD type 3 |
Autosomal recessive |
Kindlin 3 (FERMT3), involved in activation of integrin |
Kindlin 3 |
Polymorphonuclear cell, macrophage, lymphocytes, NK cells |
Tight adherence |
Same as LAD type 1 plus bleeding tendency |
Rac 2 deficiency |
Possibly autosomal dominant |
RAC2 |
Rac2, involved in regulation of actin cytoskeleton |
Polymorphonuclear cell, decreased TRECs |
Chemotaxis, O2- production |
Recurrent infections, poor wound healing, leukocytosis, poor pus formation |
E-selectin |
Possibly autosomal recessive |
Unknown |
E-selectin |
Endothelial cells |
Rolling, tethering |
Recurrent infections, poor pus formation, mild neutropenia |
NK = Natural killer, TRECs = T-cell receptor excision circles |
||||||
Molecule |
CD Number |
Distribution |
Ligand |
Function |
Integrins |
||||
LFA-1 |
CD11a/CD18 |
All leukocytes |
ICAM-1, 2, 3 |
Adhesion, migration |
CR3 |
CD11b/CD18 |
Polymorphonuclear cell, macrophage, NK cells, eosinophils |
ICAM-1,2; C3bi |
Adhesion, migration |
CR4 |
CD11c/CD18 |
All leukocytes |
C3bi, ICAM-1, CD23, fibrinogen |
Adhesion |
Alpha4-beta7 |
None |
Lymphocytes, NK cells, eosinophils |
MadCAM-1, VCAM-1, fibronectin |
Adhesion, migration, rolling |
VLA-4 |
CD49d/CD29 |
Lymphocytes, NK cells, eosinophils, basophils |
VCAM-1, fibronectin |
Adhesion, migration, rolling |
Selectins |
||||
E |
CD62E |
Endothelial cells, platelets |
Sialylated, fucosylated molecules (sLeX, CD15s) expressed on PSGL-1 and ESL-1 |
Rolling |
P |
CD62P |
Endothelial cells, platelets |
Sialylated, fucosylated molecules (sLeX) expressed on PSGL-1 |
No data |
L |
CD62L |
Leukocytes |
Sialylated, fucosylated molecules (often sulfated) expressed on CD34, MadCAM-1 and other glycoproteins-1 |
Rolling |
MadCAM = Mucosal addressin cell adhesion molecule; VCAM = Vascular cell adhesion molecule; VLA = Very late activation antigen |
||||
Subtype |
mRNA level |
CD18 Expression |
Clinical Presentation |
1 |
None |
None |
Severe |
2 |
Low |
Trace |
Moderate |
3 |
Reference range |
Trace, small protein precursor |
Moderate |
4 |
Reference range |
Large protein precursor |
Severe |
5 |
Reference range |
Normal protein precursor |
Moderate |
mRNA = messenger RNA. |
|||




