Background
Caroli disease and Caroli syndrome are rare congenital disorders of the intrahepatic bile ducts. They are both characterized by dilatation of the intrahepatic biliary tree. The term Caroli disease is applied if the disease is limited to ectasia or segmental dilatation of the larger intrahepatic ducts. This form is less common than Caroli syndrome, in which malformations of small bile ducts and congenital hepatic fibrosis are also present. This process can be either diffuse or segmental and may be limited to one lobe of the liver, more commonly the left lobe.
Caroli disease is sporadic, whereas Caroli syndrome is generally inherited in an autosomal recessive manner. [1] As with congenital hepatic fibrosis, Caroli syndrome is often associated with autosomal recessive polycystic kidney disease (ARPKD). A rare association with autosomal dominant polycystic kidney disease (ADPKD) has also been reported. See the image below.
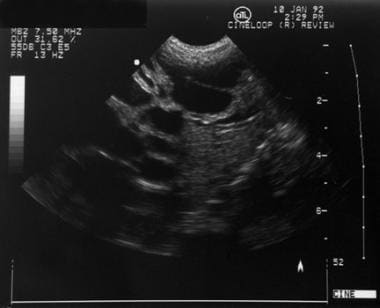 Pediatric Caroli Disease. Hepatic ultrasonogram of a neonate with Caroli disease. Multiple dilated intrahepatic bile ducts are present. Courtesy of Richard Bellah, MD, The Children's Hospital of Philadelphia.
Pediatric Caroli Disease. Hepatic ultrasonogram of a neonate with Caroli disease. Multiple dilated intrahepatic bile ducts are present. Courtesy of Richard Bellah, MD, The Children's Hospital of Philadelphia.
Pathophysiology
The precursor of the intrahepatic biliary tree is a sheath of cells surrounding the portal vein branches, known as the ductal plate (DP). The DP first arises from hepatocyte precursors surrounding hilar portal vein vessels at 8 weeks' gestation, and more peripheral regions of the DP develop sequentially. During the remainder of gestation, a process of DP remodeling occurs, during which 1-2 ductules form at points along the circumference of the DP that connect to the intrahepatic biliary tree; the remaining regions of the DP are lost, most likely through apoptosis. Caroli syndrome belongs to a subcategory of diseases thought to originate from DP malformation.
In Caroli disease, abnormalities of the bile duct occur at the level of the large intrahepatic ducts (ie, left and right hepatic ducts, segmental ducts), resulting in dilatation and ectasia. Resulting biliary stasis may lead to cholelithiasis, cholangitis, and sepsis, as well as an increased risk of cholangiocarcinoma. [1] Because reports have described cases limited to the left lobe of the liver, some have described Caroli disease as either localized or diffuse.
In Caroli syndrome, DP malformation is present at the level of the smallest portal tracts and is associated with varying degrees of portal fibrosis. These findings are typical of congenital hepatic fibrosis; therefore, Caroli syndrome is thought to belong in the same spectrum of disease as congenital hepatic fibrosis and autosomal recessive polycystic kidney disease.
Another classification of Caroli disease is type I, or simple Caroli disease, which consists of pure cystic dilatations of the intrahepatic bile ducts; type II, or complex Caroli disease, is also known as Caroli’s syndrome. [2]
Etiology
A genetic cause is likely, given the association with autosomal recessive polycystic kidney disease (ARPKD). Mutations in PKHD1 on chromosome 6p21, which is the gene linked to ARPKD, have been identified in patients with Caroli syndrome. [3, 4]
PKHD1 encodes the protein fibrocystin, which is expressed in cortical and medullary ducts in the kidney as well as biliary and pancreatic ducts in a pattern consistent with the histologic patterns seen in ARPKD.
Fibrocystin is one of a larger family of proteins that are present in the primary cilium.
Recent research supports a mechanistic link between ciliary dysfunction and polycystic kidney disease, although this remains controversial. Ciliopathies involving PCK cholangiocytes (cholangiociliopathies) in a rat model appear to be associated with decreased intracellular calcium levels and increased cAMP concentrations, causing cholangiocyte hyperproliferation, abnormal cell matrix interactions, and altered fluid secretion, which ultimately result in bile duct dilatation. [5]
The number of cases of Caroli syndrome caused by PKHD1 mutations is not known.
Caroli syndrome is inherited in an autosomal recessive manner.
Epidemiology
United States data
Caroli disease and Caroli syndrome are very rare, with an estimated incidence of less than 1 case per 100,000 population. Caroli syndrome (ectasia of the large and small bile ducts with congenital hepatic fibrosis) is more common than Caroli disease (ectasia of only the large bile ducts).
The incidence of autosomal recessive polycystic kidney disease (ARPKD)/congenital hepatic fibrosis is approximately 1:20,000 live births. [6]
Sex- and age-related data
Symptoms of Caroli disease or syndrome are more common in female patients than in male patients.
Age at presentation varies and patients may present as neonates or as adults. Cases detected in utero based on ultrasonographic findings have been reported.
Prognosis
Prognosis
Prognosis depends on the specific manifestations.
Hepatic manifestations
In the few patients who have intrahepatic ductal ectasia without associated congenital hepatic fibrosis (ie, Caroli disease), the frequency and severity of episodes of cholangitis, which may result in sepsis or death, largely determine the prognosis. Progressive liver failure may also develop, and liver transplantation may be required.
Patients with both ductal ectasia and congenital hepatic fibrosis (ie, Caroli syndrome) are subject to the risks and consequences of recurrent cholangitis, as described above. They are also at risk for the complications of cirrhosis and portal hypertension.
The risk of cholangiocarcinoma in individuals with Caroli disease or Caroli syndrome is estimated to be 100-fold higher than the general population. Cholangiocarcinoma following resection of Caroli disease or syndrome confined to a single lobe has not been reported.
Liver transplantation in patients with Caroli disease or Caroli syndrome is indicated in patients with refractory disease or with complications of hepatic fibrosis and is associated with excellent outcomes and long-term survival. Patients undergoing combined liver and kidney transplantation rather than liver transplantation alone generally have better outcomes. [7, 8]
Renal manifestations
The degree of polycystic kidney disease associated with Caroli syndrome varies. Patients who present with renal disease as neonates or infants are more likely to have severe kidney disease with enlarged cystic kidneys and progressive renal failure than others. Other patients may have normal-appearing kidneys or minimal cystic changes with only mild deficits in renal function.
Morbidity/mortality
Patients with Caroli disease or Caroli syndrome may have recurrent episodes of cholangitis and are also at risk for associated bacteremia and sepsis. Patients with Caroli syndrome or Caroli disease may have cholangitis and may also have complications of portal hypertension as is observed in congenital hepatic fibrosis. Caroli syndrome is associated with autosomal recessive polycystic kidney disease (ARPKD), and patients may have various degrees of renal cysts, interstitial fibrosis, and renal failure. Both Caroli disease and Caroli syndrome are associated with a risk of cholangiocarcinoma at a rate of 100 times that of the general population.
-
Pediatric Caroli Disease. Hepatic ultrasonogram of a neonate with Caroli disease. Multiple dilated intrahepatic bile ducts are present. Courtesy of Richard Bellah, MD, The Children's Hospital of Philadelphia.