Background
Carbamoyl phosphate synthetase (CPS) deficiency is a urea cycle defect that results from a deficiency in an enzyme that mediates the normal path for incorporation of ammonia. CPS is derived from catabolism of amino acids into a 1-carbon compound (H2 N-CO-PO32 -), in which the carbon atom is derived from bicarbonate. The process is exclusively mitochondrial and requires the expenditure of one ATP molecule. See the image below.
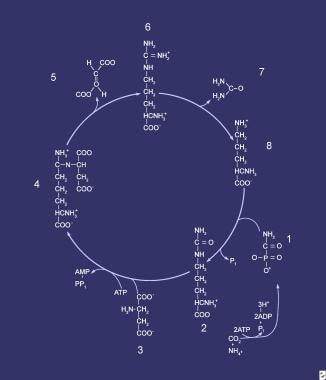 Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Pathophysiology
Two hepatocellular enzymes exist: CPS I and CPS II. CPS I is exclusively intramitochondrial, and its deficiency is responsible for the disease. CPS I is the most plentiful single protein in hepatic mitochondria, accounting for about 20% of the matrix protein. CPS II is exclusively cytosolic and is an important enzyme in de novo synthesis of pyrimidine nucleotides. The regulation of CPS I activity depends on the levels of N -acetylglutamate (see N-Acetylglutamate Synthetase (NAGS) Deficiency).
In patients with homozygous CPS I deficiency, the ability to fix waste nitrogen is completely absent, resulting in increasing levels of free ammonia with the attendant effects on the CNS. A recent molecular and functional examination of the mutational effects showed that, although some mutations affect both substrate affinity and efficiency of the reaction, others affect one more than the other. [1] Some mutations are associated with enhanced RNA instability, which leads to diminished protein synthesis. More than 190 mutations have been reported to cause CPS deficiency. [2]
The hepatic urea cycle is the major route for waste nitrogen disposal. Waste nitrogen is chiefly generated from protein and amino acid metabolism. Low-level synthesis of certain cycle intermediates in extrahepatic tissues also makes a small contribution to waste nitrogen disposal. A portion of the cycle is mitochondrial in nature; mitochondrial dysfunction may impair urea production and may result in hyperammonemia. Overall, activity of the cycle is regulated by the rate of synthesis of N -acetylglutamate, the enzyme activator of CPS I, which initiates incorporation of ammonia into the cycle.
Epidemiology
Frequency
United States
CPS deficiency is rare. As with all the urea cycle defects, as well as most of the inborn errors, citing incidence figures is impossible because new cases are generally diagnosed randomly without the benefit of population screening.
International
According a study of urea cycle diseases in Finland, 3 cases of CPS deficiency had been reported by 2007. [3] A study in Italy provided an overview of clinical findings and biochemical and molecular data concerning 13 Italian patients. [4]
Mortality/Morbidity
Mortality and morbidity rates are high. Untreated CPS deficiency is likely fatal.
Sex
CPS deficiency is autosomal recessive; thus, the incidence between the sexes is approximately equal.
Age
CPS deficiency has been reported in patients of all ages, from newborns to adults.
In adults, some individuals remain unaffected until onset in early-to-mid adulthood, whereas others gradually sustain brain damage from infancy until diagnosis.
In newborns, CPS deficiency is generally catastrophic in nature and leads to rapid demise without immediate recognition and treatment.
Prognosis
A positive outcome is extremely unlikely in individuals with neonatal onset.
Usually, the best prognosis is an infant who will be seriously impaired and will develop recurrent hyperammonemic episodes throughout infancy and childhood, sustaining further insults to the CNS.
Patient Education
As an autosomal recessive trait, each parent is assumed to be an obligate heterozygote for CPS I deficiency. The likelihood of a recurrence is 25% (1:4) with each subsequent pregnancy, regardless of fetal sex.
Prenatal diagnosis is available. If desired, contact the laboratory as early as possible.
-
Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.




