Duodenal Stenosis and Atresia
Embryology
The hepatobiliary system and pancreas form during the third week of gestation, as the second portion of the duodenum gives rise to biliary and pancreatic buds at the junction of the foregut to the midgut. The duodenum also undergoes a solid phase during this time; between 8-10 weeks' gestation, the duodenal lumen is reestablished by the gathering of vacuoles, and recanalization occurs.
Insults during this crucial period of development are believed to result in failure of recanalization and consequent atresias, stenoses, and webs. In addition, duodenal atresias have been associated with a closely surrounding piece of pancreatic tissue. Whether this tissue is an annular pancreas or merely a failure of duodenal development is debatable. [1, 2]
Classification
A stenosis is an incomplete obstruction with a small opening secondary to a diaphragm or web, whereas an atresia is a complete obstruction. Unlike other intestinal atresias, duodenal atresias are associated with other congenital anomalies. Approximately 50% of patients with duodenal atresias have some form of anomaly (eg, cardiac, anorectal, or genitourinary), and as many as 40% have trisomy 21. [3, 4, 5]
Esophageal atresia and the VACTERL (ie, vertebral defects, anal atresia, cardiac defects, tracheoesophageal fistula with esophageal atresia, renal anomalies, and limb [radial] abnormalities) syndrome have also been associated with duodenal atresia. [6] Hence, all neonates with duodenal atresia should be assessed for concomitant malformations. Growth retardation and polyhydramnios are often present antenatally.
Anatomically, most congenital duodenal obstructions are considered periampullary, with the biliary outlet occurring proximal or distal to the site of obstruction. The degree of obstruction dictates the amount of resulting pathology. The obstruction causes dilation of the proximal duodenum and stomach, as well as hypertrophy and distention of the pylorus.
A common variation is the windsock anomaly, in which the duodenum is dilated distal to the point of obstruction because of a prolapsing membrane or web (see the image below). This may be confused with a more distal duodenal obstruction.
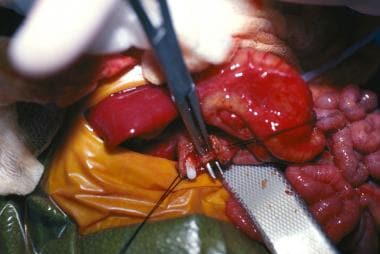
Diagnosis
Antenatal ultrasonography (US) may indicate structural and associated abnormalities, such as a dilated stomach and proximal duodenum. [7] Polyhydramnios is suggestive of a proximal gastrointestinal (GI) tract obstruction because the fetus is unable to swallow the amniotic fluid. Common associated anomalies and chromosomal defects may be assessed by screening maternal serum and amniotic fluid.
In the newborn, clear or bilious emesis is evident within hours of birth, with or without abdominal distention. An output of more than 20 mL of gastric contents is indicative of possible obstruction. [8] Because of incomplete obstruction, patients with a stenosis or web may present later with dehydration or failure to thrive.
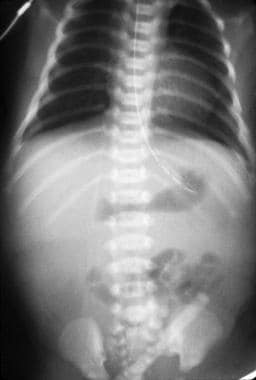
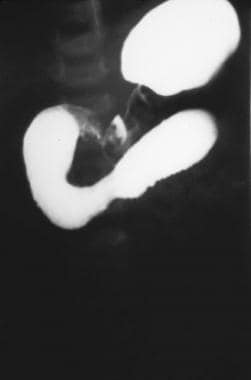
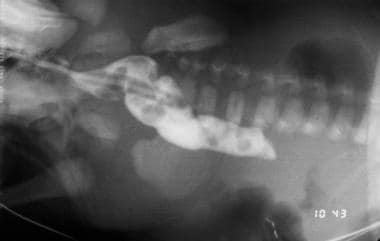
Plain radiography (see the first image below) is helpful and may reveal the classic double-bubble sign, representing air in the stomach and proximal duodenum, which is associated with complete or near-complete duodenal obstruction. Upright and contrast radiography using air or a contrast agent may confirm the diagnosis (see the second image below). Malrotation with volvulus may also result in duodenal obstruction with a double-bubble sign and can coexist with duodenal atresia in as many as 30% of cases. [9]
Treatment
Gastric decompression is essential to prevent aspiration, and thermoregulation should be monitored at all times. Unless malrotation with volvulus remains a concern, preoperative assessment of other associated anomalies should be performed. When fluid resuscitation and a full assessment have been accomplished, the neonate may proceed to surgery.
The following two basic options are available for repair of duodenal obstruction secondary to a web or atresia:
-
Duodenoduodenostomy
-
Duodenotomy with excision of the web
The most common repair is a duodenoduodenostomy. The classic approach is through a supraumbilical transverse abdominal incision that provides adequate exposure of the duodenum once the ascending and transverse colon are mobilized to the left. A catheter is inserted through the stomach to help define the site of obstruction.
In addition, the entire small bowel is carefully explored for other sites of obstruction. Care is taken to examine the gallbladder to ensure that a preduodenal portal vein is not evident and to locate the ampulla of Vater to avoid inadvertent injury. Laparoscopy has also been shown to be a safe and effective approach to repair of congenital duodenal obstruction. [10, 11, 12, 13, 14] Unlike other intestinal atresias, duodenal atresias are associated with other congenital anomalies. [15]
The proximal duodenum is usually dilated as compared with the thin-walled, flat distal duodenum and jejunum. A diamond-shaped anastomosis may be fashioned between a longitudinal incision in the distal collapsed end of the duodenum and a transverse incision in the inferior aspect of the bulbous blind-ended proximal duodenum. In the laparoscopic approach, U-clips are used for the duodenoduodenostomy. [10] The proximal duodenum is sometimes tapered by plicating its lateral antimesenteric side. A Ladd procedure is warranted if malrotation is present.
If a web is present, duodenotomy with excision of the web may be performed instead of duodenoduodenostomy. The web should be defined with the aid of a tube through the stomach, and the ampulla of Vater should be carefully identified before excision. A longitudinal incision is then made directly on the web, and the web is excised. The duodenum is then closed transversely, to avoid narrowing.
Feeding may be started when GI function is established. In some patients, this may take as long as 2 weeks. Gastric decompression via a nasogastric tube is required until that time.
Outcome
Anastomotic leakage, injury to the bile duct, and sepsis are early complications. Late complications include peptic ulceration secondary to alkaline reflux, blind-loop syndrome due to duodenal stasis, and recurrent obstruction. [16, 17, 18] In one series, as many as 12% of patients had late complications requiring reoperation for a number of reasons, including reflux and peptic ulcer disease, associated biliary abnormalities, and bowel obstruction. [9]
The prognosis is good after repair of duodenal stenosis or atresia; however, coexisting diagnoses (eg, Down syndrome and cardiac anomalies) affect outcome. [19, 20] Long-term data showed that a 9% combined early and late mortality over an average 6-year follow-up period was due almost exclusively to associated congenital anomalies. [9] In addition, birth weight is a predictor of mortality. Infants with duodenal atresia who weigh less than 2 kg at birth have poorer survival, regardless of the presence of other congenital abnormalities. [21]
Jejunoileal Stenosis and Atresia
Etiology
Multiple theories regarding the etiology of jejunoileal atresia have been studied in many animal models (eg, puppies, ewes, rabbits, and chick embryos). [22, 23, 24, 25, 26] Murine studies suggest that some forms of atresia may be hereditary and result from dysregulation of proliferation and apoptosis of the developing intestine through the fibroblast growth factor pathways. [27] To date, the best-accepted etiologic theory has been that of an intrauterine vascular accident resulting in necrosis of the affected segment, with subsequent resorption. [4, 23]
Classification
Jejunoileal defects may be divided into two broad categories, stenosis and atresia. A stenosis has an intact mesentery and is a localized narrowing of the bowel. Luminal continuity is not lost, and the stenotic portion of the bowel generally has an irregular muscularis and a thickened submucosa. Four types of jejunoileal atresias are described, [4, 23, 28, 29] representing a spectrum of severity ranging from a simple web to full atresia with loss of bowel length. This classification generally guides prognosis and therapy (see the image below).
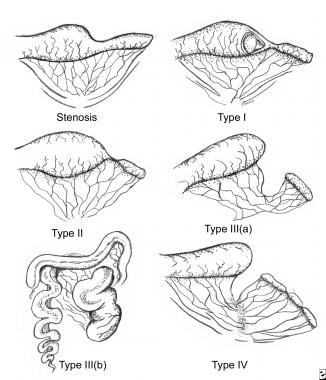
Atresia types
In type I jejunoileal atresias, the mucosa and submucosa form a web or intraluminal diaphragm, resulting in obstruction. As in duodenal webs, a windsock effect may be evident secondary to an increase in intraluminal pressure in the proximal bowel causing a prolapse of a portion of the web into the distal part of the bowel. A mesenteric defect is not present, and the bowel length is not affected.
In type II atresias, the mesentery is intact; however, the bowel is not joined. The dilated proximal portion has a bulbous blind end connected by a fibrous cord to the blind end of the distal flattened bowel. The overall length of the small bowel is not usually shortened.
The defect in type IIIa atresias is similar to that in type II atresias, in that both types have blind proximal and distal ends; however, in type IIIa, no bridging fibrous cord is present, and a V-shaped mesenteric defect is present. The proximal blind end is usually markedly dilated and aperistaltic. The intervening bowel has undergone intrauterine resorption, and, as a result, the bowel in this category is variably shortened.
In type IIIb atresias, besides a large mesenteric defect, significant bowel shortening is noted. This lesion is also known as a Christmas tree or apple-peel deformity, because of the bowel's appearance as it wraps around a single perfusing vessel. [30, 31] The distal small bowel receives its blood supply from a single ileocolic or right colic artery because the better part of the superior mesenteric artery is absent. This deformity has been linked to prematurity, malrotation, and subsequent short-bowel syndrome, with increased morbidity and mortality. [32, 33]
A type IV atresia involves multiple small-bowel atresias of any combination of types I to III. This defect often takes on the appearance of a string of sausages because of the multiple lesions. The cause is unknown, and etiologic theories range from multiple ischemic infarcts, possibly from a more global placental insufficiency, to an early embryologic defect of the GI tract, to an inflammatory process occurring in utero. [26, 28, 34]
Diagnosis
Antenatal diagnosis of jejunoileal lesions by means of US and antenatal screening is possible; however, the reported accuracy of antenatal US in this setting has varied widely, and there is a need for better diagnostic criteria. [35] Whereas associated anomalies are found in 30-40% of neonates with duodenal atresias, associated anomalies are found in only 10% of neonates with jejunoileal atresias. [6]
Clinically, neonates with a proximal atresia develop bilious emesis within hours, whereas patients with more distal lesions may take longer to begin vomiting. A normal or scaphoidlike abdomen in a neonate with bilious emesis should be considered indicative of a proximal obstruction until proven otherwise. Abdominal distention is more pronounced with distal lesions.
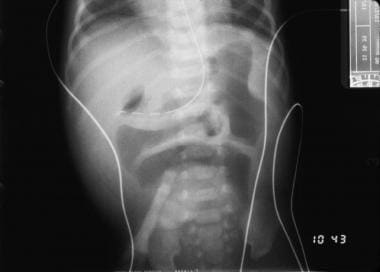
Radiography is helpful for confirming the diagnosis. With more proximal atresias, few air-fluid levels are evident with no apparent gas in the lower part of the abdomen. The more distal lesions demonstrate more air-fluid levels, though the distal intestine remains gasless (see the image below).
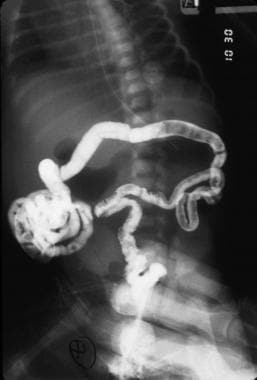
Although plain radiography can depict the presence of an obstruction, it is not the best method of showing the location of the abnormality. A barium enema may be used to define a microcolon indicative of a distal small-bowel obstruction; it is also capable of establishing the diagnosis of other causes of lower obstruction, such as Hirschsprung disease or a meconium plug. The contrast enema may also reflux into the small bowel and help define the level of a distal obstruction (see the images below).
Stenosis in a neonate is more difficult to diagnose, and it may not manifest for some time. The clinical presentation is dependent on the severity of disease, and these patients have a history of intermittent emesis and failure to thrive. An upper GI series with small-bowel follow-through is indicated in these patients.
Treatment
Preoperative care should be the same as that described for repair of duodenal lesions. Once the diagnosis is made, the patient should be fully resuscitated before surgical correction is attempted, unless a perforation or volvulus is suspected. Gastric decompression, fluid resuscitation, and thermoregulation are essential. Preoperative antibiotics should be administered.
In the operating room, a transverse supraumbilical incision affords adequate exposure of the abdominal contents. The full length of the bowel is manually explored for malrotation, other atresias, or stenoses; care is taken to keep the bowel moist and protected at all times. Malrotation should be treated with a Ladd procedure.
The length of intestine that appears functional is measured along the antimesenteric border because bowel length affects the procedure and overall prognosis. Sodium chloride solution is injected into the distal bowel and followed closely to the cecum to ensure patency. The same is done for the colon.
Once patency of the entire length of the bowel is established, the repair may proceed. The dilated proximal bulb generally does not have normal function; consequently, it should be resected up to a more suitable size to avoid problems with abnormal peristalsis postoperatively. If the bowel length is limited, a tapering enteroplasty should be considered rather than resection. An end-to-end anastomosis can then be performed (see the images below).
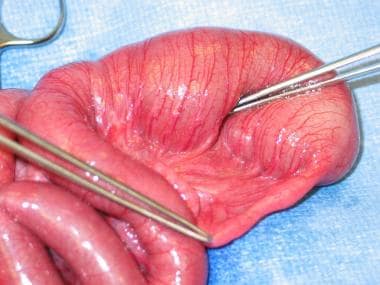 Type I jejunal atresia with intact serosa and mesentery. Image courtesy of Eugene S Kim, MD.
Type I jejunal atresia with intact serosa and mesentery. Image courtesy of Eugene S Kim, MD.
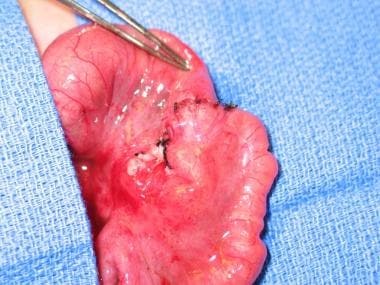 Type I jejunal atresia with intact serosa and mesentery. Resection and end-to-end anastomosis of atresia. Image courtesy of Eugene S Kim, MD.
Type I jejunal atresia with intact serosa and mesentery. Resection and end-to-end anastomosis of atresia. Image courtesy of Eugene S Kim, MD.
An alternative technique, serial transverse enteroplasty, has been used to both taper and lengthen the bowel in neonates with atresia and shortened bowel. [36, 37] In addition, in the specific case of type IV atresia with minimal remnant bowel, creative techniques such as stenting of multiple bowel segments have been performed with some success. [38]
Postoperatively, gastric drainage should continue until the return of bowel function. Total parenteral nutrition (TPN) should be started shortly after surgery and continued until enteral feeds are tolerated. The baby must be closely monitored after surgery for abdominal girth, vital signs, temperature, fluid status, urine output, gastric drainage, and level of activity.
Outcome
In general, children with atresia do well postoperatively. Type IIIb atresia, by its nature, has a relatively poor prognosis, though in one long-term series, even these children had a relatively low rate of late morbidity. [39] As with duodenal atresias, mortality is primarily associated with the presence of other congenital anomalies, as well as birth weight. [21]
Dysmotility of the remnant bowel near sites of atresia does occur, as mentioned above, and may be related to a dysgenesis of enteric neuronal structures. [40] If the bulbous portions of atretic bowel are not resected because of limited bowel length, this dysmotility can cause late complications of stasis, bacterial overgrowth, and feeding intolerance.
The overall prognosis for patients with jejunoileal atresia depends on the amount of functional small bowel remaining after surgery. In general, 40 cm of functional bowel is considered adequate, even without an ileocecal valve; however, neonates with as little as 10 cm of small bowel have been successfully weaned from parenteral nutrition. [41]
Children with jejunoileal atresias take longer to reach full enteral nutrition than those with either duodenal or colonic atresias do, with an average time to enteral autonomy of greater than 2 weeks. [21] For optimal results, these neonates with intestinal inadequacy require careful follow-up care with surgeons, pediatric gastroenterologists, and dietitians.
Colonic Atresia
Etiology and classification
Like jejunoileal atresia, colonic atresia (see the images below) is believed to be caused by a vascular accident in utero that results in ischemic injury, probably after the midgut has returned to the coelomic cavity. Some animal evidence, as discussed above, ties colonic atresia to a heritable defect in the fibroblast growth factor regulatory pathway. [27, 42]
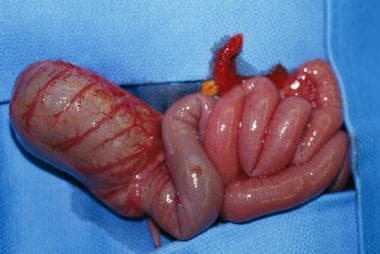
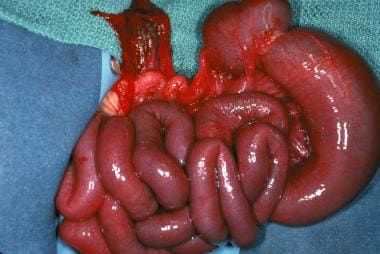
Colonic atresia is the least common of all intestinal atresias and stenosis, accounting for only 1.8-15% of such defects. [43, 44] Atresias may occur throughout the colon; however, lesions proximal to the splenic flexure and distal to the vascular watershed area are the most common. [43] Colonic atresias are frequently associated with other anomalies, including jejunoileal atresia, Hirschsprung disease, and genitourinary malformations. [45] They can generally be classified in the same fashion as duodenal or jejunoileal atresias.
Diagnosis
Antenatal US may detect a colon larger than that expected for gestational age in patients with colonic atresia. [46] Diagnosis after birth is usually timely because the newborn demonstrates signs of distal bowel obstruction. Abdominal distention is prominent in the first 24 hours, and the hugely dilated proximal loop is often palpable.
Radiographs depict a large loop of bowel with proximal air-fluid levels. A contrast enema can provide a definitive diagnosis and supply anatomic information about the location of the lesion (see the image below). This study can usually be used to differentiate colonic atresia from meconium ileus, Hirschsprung disease, and other intestinal atresias.
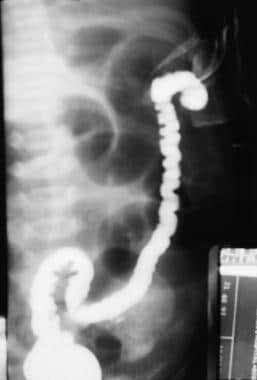
Treatment
Treatment depends on the extent and location of the lesion and the clinical presentation of the patient. Care should be taken to avoid perforation secondary to severe distention. A staged procedure, beginning with resection of the affected portion and colostomy with mucous fistula, is generally the preferred initial treatment of choice because of the extreme dilation of the proximal colon. Ileocolic or colocolic anastomosis should be performed as a second procedure.
In addition, care must be taken to rule out other anomalies, including multiple atresias and Hirschsprung disease. [47]
Outcome
Outcome is dependent on associated anomalies, including small-bowel atresias and birth weight. [21] The prognosis is generally good.
-
Duodenal web windsock deformity.
-
Contrast study illustrating duodenal stenosis.
-
Plain radiograph of duodenal stenosis.
-
Classification of jejunoileal atresias.
-
Plain radiograph demonstrating jejunal atresia.
-
Contrast study of jejunal atresia.
-
Contrast study of jejunal atresia.
-
Type I jejunal atresia with intact serosa and mesentery. Image courtesy of Eugene S Kim, MD.
-
Type I jejunal atresia with intact serosa and mesentery. Resection and end-to-end anastomosis of atresia. Image courtesy of Eugene S Kim, MD.
-
Contrast study of colonic atresia.
-
Gross example of colonic atresia.
-
Gross example of colonic atresia.


