Practice Essentials
Congenital abnormalities of fibrinogen are divided into two types: type I, or quantitative abnormalities (afibrinogenemia and hypofibrinogenemia), and type II, or qualitative abnormalities (dysfibrinogenemia and hypodysfibrinogenemia). Afibrinogenemia and hypofibrinogenemia are quantitative defects in fibrinogen (type I), which result from mutations that affect plasma fibrinogen concentration inherited on both chromosomal alleles and are frequently associated with a bleeding diathesis but occasionally a thrombotic event. [1] Dysfibrinogenemia is a qualitative defect in fibrinogen (type II) marked by functional abnormalities of fibrinogen who carry one abnormal allele that may result in either bleeding or thrombosis. [2, 3, 4, 5]
Fibrinogen is a 340-kD glycoprotein that is synthesized in the liver and circulates in plasma at a concentration of 2-4 g/L, with a half-life of 4 days. The fibrinogen molecule is a hexamer, consisting of 3 paired polypeptide chains: A-α, B-β, and γ; A and B refer to specific polypeptides on 2 of the chains. Synthesis of the protein in hepatocytes is under the control of 3 genes (one for each chain), FGA, FGB, and FGG, located within 50 kilobases (kb) on chromosome 4 (4q).
The primary physiologic role of fibrinogen is in hemostasis. In the final step of the coagulation cascade, fibrinogen is converted to fibrin, with formation of a fibrin clot. The first step in this conversion is thrombin cleavage of fibrinopeptides A and B from the fibrinogen α and β chains; the residual molecule is referred to as fibrin monomer. A loose fibrin clot develops as fibrin monomers spontaneously polymerize. The formation of a firm insoluble fibrin gel depends on cross-linking of the polymer by the transglutaminase activity of factor XIIIa (see the image below).
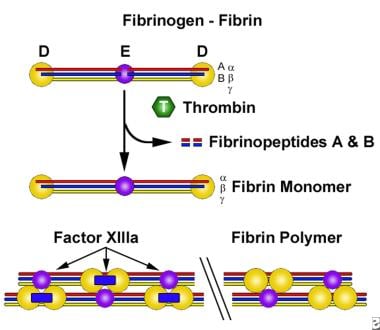 The conversion of soluble fibrinogen to insoluble fibrin.
The conversion of soluble fibrinogen to insoluble fibrin.
The fibrin clot has an essential role in limiting bleeding at sites of blood vessel injury; it also provides the structure for assembly and activation of the fibrinolytic proteins.
Although the primary function of fibrinogen is in fibrin clot formation, it has a multitude of other functions, including nonsubstrate thrombin binding, platelet aggregation, and fibrinolysis. Exposure of its nonsubstrate thrombin-binding sites after fibrin clot formation promotes the antithrombotic properties of fibrinogen. [6] Therefore, disorders of fibrinogen may be associated with either a bleeding or a thrombotic predisposition.
Signs of fibrinogen abnormalities
Umbilical cord hemorrhage frequently provides an early alert to afibrinogenemia. Other bleeding manifestations include the following:
-
Epistaxis and oral mucosal bleeding
-
Hemarthrosis and muscle hematoma
-
Gastrointestinal (GI) bleeding
-
Menorrhagia and postpartum hemorrhage
-
Traumatic and surgical bleeding
-
Spontaneous splenic rupture and intracranial hemorrhage (rare) [7]
In patients with hypofibrinogenemia, bleeding episodes are usually mild, and, in many cases, no spontaneous clinical bleeding is present; bleeding may occur following trauma or surgery. [8, 9]
Afibrinogenemia and hypofibrinogenemia can be associated with thrombosis and with recurrent spontaneous abortion.
Patients with dysfibrinogenemia may experience hemorrhage or thrombosis, but most are asymptomatic. [10]
Workup in fibrinogen abnormalities
Screening tests
The prothrombin time (PT) and activated partial thromboplastin time (aPTT) are prolonged in afibrinogenemia and may be prolonged in hypofibrinogenemia and dysfibrinogenemia. However, these tests have a poor sensitivity to mild fibrinogen deficiency or dysfunction.
Thrombin time is more sensitive than PT or aPTT for quantitative and qualitative defects in fibrinogen. However, the specificity is poor because a prolonged thrombin time can occur in the presence of heparin, a high concentration of fibrin degradation products (FDPs), and direct thrombin inhibitors. Furthermore, results can significantly vary between laboratories as the test is not standardized.
A snake venom that directly activates fibrinogen by cleaving fibrinopeptide A is used as a reagent in the reptilase time test. The advantage over the thrombin time is that this test is not affected by heparin. A prolonged reptilase time, in the presence of a normal fibrinogen concentration, provides strong evidence of a dysfibrinogenemia. However, this test does not detect all forms of dysfibrinogenemia.
Clottable fibrinogen
A functional assay by the Clauss method is one of the most common tests used to measure fibrinogen activity. In this method, a reagent containing a high concentration of thrombin that triggers clot formation when added to citrated plasma is used.
Fibrinogen antigen
In afibrinogenemia, fibrinogen concentrations are low using the clottable or quantitative antigen method.
In dysfibrinogenemia, a discrepancy may be found between fibrinogen measured in a functional assay (low) and fibrinogen measured immunologically (normal); however, in some dysfibrinogenemias, a concordant decrease in the two assays is observed.
Genotyping
Genotyping identification of the specific molecular defect may be useful for diagnosis confirmation in afibrinogenemia and dysfibrinogenemia.
Imaging studies
In the investigation of suspected bleeding, appropriate imaging studies (eg, brain computed tomography [CT] scanning or magnetic resonance imaging [MRI]) may reveal the presence of suspected central nervous system (CNS) hemorrhage.
Management of fibrinogen abnormalities
Medical care in fibrinogen abnormalities can include the following:
-
Hemorrhage - Fibrinogen administration may offer a prophylactic benefit to patients with afibrinogenemia
-
Thrombosis - Patients who present with thrombosis associated with dysfibrinogenemia should receive anticoagulation therapy
-
Spontaneous abortion - Recurrent spontaneous abortion may be prevented by routine prophylaxis with fibrinogen concentrates starting early in pregnancy
Pathophysiology
Mutations impacting fibrinogen synthesis or processing give rise to quantitative fibrinogen deficiencies, while mutations causing abnormal polymerization, cross-linking, or assembly of the fibrinolytic system lead to qualitative defects. [9] Genetic defects and pathogenic mechanisms that have been identified include deletions, point mutations resulting in premature termination codons, missense mutations disturbing fibrinogen assembly or secretion, and unilateral isodisomy related to a significant deletion. Although mutations have been found in all three of the fibrinogen genes, the most common defects are aberrant splicing and deletion mutations in the fibrinogen A gene. Mutation-related molecular defects, identified through studies of specific mutations, include truncated α or γ chains or aberrantly folded β chains. Mutations can interfere with peptide synthesis or assembly of the fibrinogen hexameric complex and its secretion from the hepatocyte. [11, 12] These disorders are usually diagnosed in the newborn period, when they can present with umbilical cord bleeding.
Congenital dysfibrinogenemia is the result of mutations that give rise to functional abnormalities. The presence of an associated bleeding tendency or an increased risk of thrombosis depends on the effect of the specific mutation.
Type I (quantitative) fibrinogen deficiencies are generally inherited as autosomal recessive traits, whereas type II (qualitative) dysfibrinogenemias are inherited as autosomal dominant disorders in most cases.
Mutations associated with bleeding
There is a strong correlation between fibrinogen activity level and severity of bleeding. Abnormalities at the thrombin cleavage site of the Aα chain result in impaired release of fibrinopeptide A, inhibiting the conversion of fibrinogen to fibrin. Absent or slow fibrinopeptide release with delayed polymerization of the fibrin monomers has been associated with mutations in all 3 of the fibrinogen genes. Abnormal fibrinogens that exhibit defective cross-linking by factor XIIIa have been associated with abnormal wound healing.
In a study of 102 patients with congenital dysfibrinogenemia, Zhou et al found bleeding in 27.5% of them and thrombosis in 3.9%, while 68.6% of patients were asymptomatic. Thromboelastography results differed significantly between patients with hot-spot mutations at AαArg35(16) and γArg301(275), although such differences were not found between patients with and without bleeding. Thromboelastography results were normal in patients with mutations at AαArg35(16), AαPro37(18), or AαArg38(19). [13]
Mutations associated with thrombosis
Impaired fibrinopeptide B release results in abnormalities of polymerization that are associated with thrombotic events.
Abnormalities that interfere with plasminogen binding or activation on the fibrin clot result in reduced fibrinolysis and are associated with clinical thrombosis.
Defective fibrin binding of thrombin (a process that normally limits thrombin activity) results in prolonged activity of unbound thrombin, leading to amplification of fibrin clot formation and enhanced platelet activation. Mutations may be clinically silent.
Epidemiology
Frequency
United States
A North American Registry of Rare Bleeding Disorders has been successful in collecting valuable information on inherited fibrinogen disorders and other rare bleeding disorders, with respect to disease prevalence, genotyping frequency, diagnostic events, clinical manifestations, treatment, and prophylaxis strategies, as well as disease and treatment-related complications. Among all the reported cases of fibrinogen disorders in this registry, afibrinogenemia accounted for 24% of cases, hypofibrinogenemia accounted for 38%, and dysfibrinogenemia accounted for 38%. [14] More recently, other resources for clinicians include the Rare Coagulation Disorders Resource Room.
International
The frequency of afibrinogenemia is estimated to be 1-2 cases per million people; a high rate of consanguinity has been reported. Inherited dysfibrinogenemia in the general population is rare, but determination of the true incidence is difficult because many patients are asymptomatic. In addition to the North American Registry, several other recent registries from Italy, Iran, and the United Kingdom have greatly improved understanding of the clinical spectrum of presentation. In one large registry of cases, at least half of the patients were asymptomatic. [15] Less than 1% of patients with venous thrombosis who were evaluated for dysfibrinogenemia were found to have this abnormality. A registry in Europe is also collecting data on this rare disorder.
Mortality/Morbidity
Deaths attributable to afibrinogenemia are associated with bleeding, most commonly postoperative bleeding and intracranial hemorrhage. Recurrent spontaneous abortions can occur in women with afibrinogenemia. Patients with dysfibrinogenemia are at risk of bleeding or thrombosis. [16]
Sex
Afibrinogenemia is autosomal recessive, with a male-to-female ratio of 1:1. Dysfibrinogenemias may manifest either autosomal recessive or autosomal dominant inheritance.
Afibrinogenemia poses a major risk during pregnancy and after delivery. Indeed, dysfibrinogenemia and thrombosis may be overrepresented in women because of the increased risk of thrombosis associated with pregnancy and the postpartum period.
Age
The age at diagnosis varies. Afibrinogenemia is often first diagnosed in the newborn period because of umbilical cord bleeding. [17]
Hypofibrinogenemia (ie, less severely reduced fibrinogen levels) is associated with fewer bleeding episodes and may be first diagnosed at the time of a traumatic or surgical challenge that results in bleeding.
Dysfibrinogenemias are commonly diagnosed in adulthood.
-
The conversion of soluble fibrinogen to insoluble fibrin.


