Practice Essentials
Colorectal cancer is uncommon in adolescents and young adults; nonetheless, the incidence of pediatric colorectal tumors is rising. This trend is discordant with that seen in older adults, who are benefiting from screening programs. [1] The incidence of distal colon and rectal tumors is rising at the fastest rate, and rectal tumors are disproportionately represented in the very young age groups.
Most cases of colorectal cancer in adolescents and young adults are sporadic, but several genetic syndromes are associated with these tumors in young patients. The differing biology is suggested by the preponderance of high-grade and mucinous tumors, but the unique oncogenesis is not fully understood. See the images below.
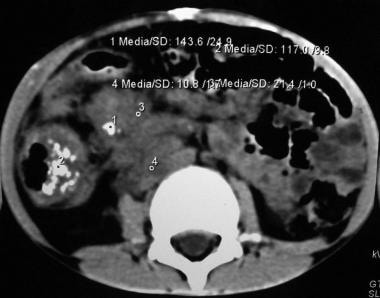 This picture depicts an abdominal CT scan of a 7 year-old boy with a mucinous adenocarcinoma of the ascending colon. Note the thickness and increased vascularity of the colonic wall, as well as irregularities on the serosal surface. This cut also shows severe tumor infiltration of the colonic mesentery surrounding the mesenteric and retroperitoneal vessels.
This picture depicts an abdominal CT scan of a 7 year-old boy with a mucinous adenocarcinoma of the ascending colon. Note the thickness and increased vascularity of the colonic wall, as well as irregularities on the serosal surface. This cut also shows severe tumor infiltration of the colonic mesentery surrounding the mesenteric and retroperitoneal vessels.
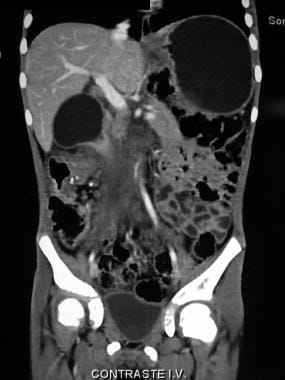 Coronal CT scan demonstrating the profuse tumoral infiltration of the ascending colonic mesentery surrounding mesenteric and portal vessels. Also note the thickness of the colonic hepatic flexure.
Coronal CT scan demonstrating the profuse tumoral infiltration of the ascending colonic mesentery surrounding mesenteric and portal vessels. Also note the thickness of the colonic hepatic flexure.
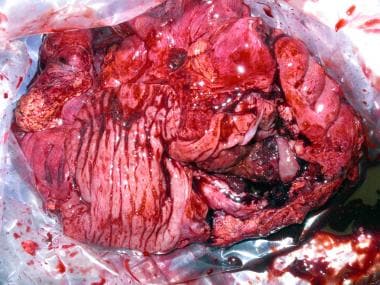 Surgical specimen after right hemicolectomy, including the terminal ileum up to the transverse colon. Mesenteric fat, vessels and lymph nodes were resected en block with the ascending colon. The large intestine has been opened longitudinally. Note the tumor on the right lower quadrant of the image, with severe thickness of the wall, areas of necrosis and hemorrhage, and some stippled calcifications.
Surgical specimen after right hemicolectomy, including the terminal ileum up to the transverse colon. Mesenteric fat, vessels and lymph nodes were resected en block with the ascending colon. The large intestine has been opened longitudinally. Note the tumor on the right lower quadrant of the image, with severe thickness of the wall, areas of necrosis and hemorrhage, and some stippled calcifications.
Multidisciplinary management, including medical, radiation, and surgical oncology teams, is crucial for optimal outcomes.
Surgical treatment of metastatic colorectal cancer offers a chance for cure or prolonged survival.
Patients with peritoneal metastatic disease may benefit from surgical cytoreduction and hyper-thermic intraperitoneal chemotherapy (HIPEC). Best outcomes are documented for those in whom complete cytoreduction is surgically feasible.
Pulmonary metastases are rarely isolated; however, surgical resection is associated with prolonged survival in selected patients. [2]
Background
Fewer than 1% of cases of adenocarcinoma of the colon and rectum occur in patients younger than 20 years of age. [1] Late diagnosis is not uncommon because of the rarity of this entity and the failure to include it within the differential diagnosis for rectal bleeding, chronic abdominal pain, or bowel obstruction in children.
Colorectal cancer (CRC) is the most common cancer of the gastrointestinal (GI) tract. In adults, it represents the third most common cancer overall, and the second leading cause of cancer-related deaths. In the United States, about 150,000 new cases are diagnosed annually, with only a minority of cases (approximately 80 per year [< 1%]) being diagnosed in adolescents and young adults, aged 15-39 years. [3] In recent decades, effective screening has reduced the incidence of and mortality from CRC. However, young-onset CRC is on the rise.
CRC linked to familial syndromes is more common among adolescents and young adults; however, the vast majority of cases in this age group are sporadic. More recent literature has focused on age-based disparities related to CRC. [3]
See Benign or Malignant: Can You Identify These Colonic Lesions?, a Critical Images slideshow, for characteristic features of benign lesions as well as those with malignant potential.
Polypoid Disease of the Gastrointestinal Tract
Not all polyposis syndromes are familial. Familial polyposis syndromes are divided into 2 major groups based on the presence of adenomas or hamartomas. The inherited adenomatous polyposis syndromes include familial adenomatous polyposis (FAP) and Turcot syndrome; the familial hamartomatous polyposis syndromes include Peutz-Jeghers syndrome and juvenile polyposis.
Although juvenile polyps are common in children, adenomas are quite unusual. The latter are considered dysplastic precancerous lesions that are commonly seen in late adulthood. When discovered in children, they suggest one of several types of inherited CRC. [4]
Although the nomenclature is confusing, diffuse juvenile polyposis differs from juvenile polyposis coli. Diffuse juvenile polyposis is a syndrome with multiple polyps spread throughout the GI tract and presents in younger children (aged 6 months to 5 years); in juvenile polyposis coli, the polyps are confined to the rectosigmoid area and are typically found in older patients (aged 5-15 years). Hamartomatous polyps may also be found in patients with Cowden disease, Cronkhite-Canada syndrome, Bannayan-Riley-Ruvalcaba syndrome, and basal cell nevus syndrome. [5]
Colonic polyposis syndromes
Colonic polyposis syndromes include the following:
-
Nonfamilial polyposis - Isolated juvenile polyps (inflammatory polyps)
-
Familial polyposis - Adenomas (FAP, Gardner syndrome, Turcot syndrome) and hamartomas (juvenile polyposis, Peutz-Jeghers syndrome, Cowden disease, Cronkhite-Canada syndrome)
The lesions can be isolated to the intestine (eg, juvenile, lymphoid, familial adenomatous) or can involve other areas of the body (eg, Peutz-Jeghers syndrome, Gardner syndrome, Turcot syndrome). Most polyps of the GI tract are benign and result from hamartomas of the mucosa or lymphoid hyperplasia of the submucosal layer. However, adenomatous polyps represent a genetic alteration in the mucosa and have substantial malignant potential.
For study purposes, only the hamartomatous lesions and other nonfamilial lesions are discussed in this section. FAP is presented in detail below, with other cancer-predisposing entities.
Polyps occur in 1% of preschool-aged and school-aged children and are the most frequent cause of rectal bleeding in toddlers and infants aged 2-5 years. Juvenile polyps are the most common (80%), followed by lymphoid polyps (15%). [6]
Isolated juvenile polyps (nonmalignant) involve no family history of juvenile polyposis and fewer than 5 polyps confined to the colon.
Juvenile polyposis syndromes with malignant potential are classified as follows [7] :
-
Diffuse juvenile polyposis of infancy - Widespread polyposis of the entire GI tract in patients younger than 6 months.
-
Diffuse juvenile polyposis - Multiple polyps throughout the GI tract but concentrated in the stomach, distal colon, and rectum; usually occurs in patients aged 6 months to 5 years.
-
Juvenile polyposis coli - Multiple polyps confined to the distal colon and rectum in patients aged 5-15 years.
Lymphoid polyps (lymphoid nodular hyperplasia)
Lymphoid polyps (present in 15% of patients) are hyperplastic submucosal lymphoid aggregates, most likely due to a nonspecific infection (exposure to bacteria and viruses). Submucosal lymphoid tissue is prominent in children, particularly in the distal ileum (Peyer patches). These non-neoplastic polyps may occur in the rectum, colon, and terminal ileum.
Macroscopically, they appear as firm, round, submucosal nodules that are smooth or lobulated. They are never pedunculated. They often have a volcano-like appearance with mucosal ulceration, which leads to occult blood loss. Histologically, they are hyperplastic lymphoid follicles with a large germinal center covered by colonic mucosa. They develop in young children, with a peak incidence at age 4 years.
Patients present with anemia or, less frequently, with severe rectal bleeding. Barium enema and colonoscopy findings are helpful (in 50% of patients), and biopsy findings confirm the diagnosis.
Surgery is indicated only for uncontrolled bleeding and intussusception that does not respond to enema treatment. Otherwise, expectant measures are adequate because these polyps are benign and spontaneously regress.
Isolated juvenile polyps
Juvenile polyps are mucosal tumors that consist of excessive lamina propria and dilated cystic glands. They usually occur between 2 and 10 years of age and constitute 80% of all polyps in children, with a slight male predominance (3:2). [8]
Juvenile polyps typically present as painless rectal bleeding after defecation. A small percentage debut with prolapse. Prolapsed polyps appear as dark, beefy-red, pedunculated masses, in contrast to the lighter pink mucosal appearance of rectal prolapse. Most prolapsed polyps are erythematous and friable. Abdominal pain is uncommon and is primarily associated with intussusception. [9]
Colonoscopy is the procedure of choice for evaluation of juvenile polyps because those identified in the distal GI tract can be removed at the same time.
Juvenile polyps are solitary in half of patients. The majority are located proximal to the rectosigmoid junction. However, those distally located tend to cause more symptoms. Removal for histologic confirmation is indicated. The typical isolated juvenile polyp with no adenomatous changes has no potential for malignancy and tends not to recur.
Juvenile polyposis syndromes
Juvenile polyposis syndrome (JPS) is a genetic disorder associated with an increased risk of colorectal cancer. It is diagnosed in patients with more than 5 polyps at the colon/rectum, multiple polyps throughout the upper and lower GI tract, or any number of lesions with a family history of juvenile polyposis.
Surgery may be offered when the patient has too many polyps to treat endoscopically, or for patients with GI bleeding, anemia, diarrhea, or protein-losing enteropathy. [8]
Diffuse juvenile polyposis of infancy
This entity occurs within the first months of life and is not familial. Patients may present with diarrhea, rectal bleeding, intussusception, prolapse, bowel, protein-losing enteropathy, macrocephaly, clubbing of fingers and toes, and hypotonia. [10]
The entire GI tract is involved. One third of these patients have other congenital abnormalities such as Meckel diverticulum, malrotation, and heart lesions. [7]
Patients require total parenteral nutrition (TPN) and bowel rest, followed by selective resection.
Despite appropriate treatment, this disease is almost universally fatal; only 2 patients have been reported to survive after age 2 years. [10]
Diffuse juvenile polyposis
Diffuse or familial juvenile polyposis was originally identified as isolated or multiple hamartomatous polyps that occur in the colon and rectum of children aged 6 months to 5 years.
Patients present with bright red blood per rectum, anemia, abdominal pain, and rectal prolapse. Diffuse juvenile polyposis is inherited as an autosomal dominant trait [7] ; thus, if a parent has the condition, the risk of having an affected child is 50%.
Hamartomas are malformed colonic mucosa arranged in a bizarre fashion. Typically, hamartomas are not considered premalignant unless they are part of a polyposis syndrome.
Patients with diffuse juvenile polyposis have a 50% lifetime risk of colorectal carcinoma. This may be due to chronic inflammation that produces reactive hyperplasia, which then progresses to dysplasia or adenomatous changes. These polyps often have an ulcerated surface and demonstrate more epithelium with a villous or papillary configuration.
In addition to the epithelial dysplasia that occurs in juvenile polyps, adenomas are also often present. Thus, the approach to affected patients is similar to that taken in patients with FAP. Some authors recommend monitoring these patients with an annual complete blood cell (CBC) count (to detect anemia due to GI bleeding), semiannual colonoscopy, and subsequent colectomy if severe dysplasia, bleeding, or rapid polyp formation occurs. Others advocate for prophylactic colectomy.
Associated congenital defects include cleft palate, malrotation, polydactyly, and cranial abnormalities.
Juvenile polyposis coli
A child with 3-10 colonic polyps, any number of polyps in the GI tract outside of the colon, or one polyp and a family history of juvenile polyposis is considered to have the syndrome.
Most patients have 50-100 colorectal polyps; they may also have gastric and small intestinal polyps.
Identifying patients with this syndrome is fundamental because of the high risk for carcinoma (17%) at an early age; the mean age at diagnosis of carcinoma is 35.5 years. [11]
Close long-term surveillance is important. The amount of polyps increases the risk of chronic bleeding, which subsequently leads to iron deficiency anemia, hypoproteinemia, and failure to thrive. [12, 13]
Macroscopically, these polyps resemble the isolated juvenile polyps; however, histologically, they have more epithelium with a villous or papillary configuration. Epithelial dysplasia can occur. Adenomas can also be found in conjunction with juvenile polyps. [10] Lobular polyps have a higher propensity for a more severe dysplasia (47%) than non-lobular polyps (10%). [14]
According to the St. Mark's Polyposis Registry in London, the cumulative risk of cancer in patients with a juvenile polyposis syndrome is 68% by age 60 years. [15] Because the entity is transmitted in an autosomal dominant fashion, patients with a juvenile polyposis syndrome and their families must receive long-term follow-up. [16]
Some authors advocate prophylactic total colectomy and rectal mucosectomy with an endorectal pull-through (ERPT), [14] whereas others recommend regular screening with colonoscopy and subsequent colectomy if severe dysplasia, rapid polyp formation, or bleeding occurs. [17]
When intussusception occurs in children older than 2 years, the discovery of a specific lead point is not uncommon (22%); however, lead points are only found in 2-8% of children within the usual age range (6-18 months). [9] When a polyp is demonstrated as a lead point in a patient with intussusception, an evaluation may be indicated to identify polyposis syndromes.
Some hamartomas do not appear to have any malignant potential. However, germline mutations and somatic inactivation of STK11, SMAD4, BMPR1A, and PTEN genes in hamartomatous polyposis syndromes create an epithelial environment favorable for neoplastic transformation. [4]
Peutz-Jeghers syndrome
Peutz-Jeghers syndrome is a cancer-predisposing disorder that is inherited as an autosomal dominant trait. It is characterized by mucocutaneous pigmentation and hamartomatous polyps of the GI tract. Since the tumors tend to recur, extensive bowel resection should be avoided to prevent short bowel syndrome. [18]
In 1921, Peutz reported on the association of intestinal polyps with mucocutaneous pigmented spots of the mouth, hands, and feet. [19] From 1944-1949, in a study of 20 patients, Jeghers defined the 2 main features of the syndrome as melanotic spots on the buccal mucosa and lips (with variable melanin pigmentation on the face and digits) and polyposis of the intestinal tract. [10] The melanotic spots range from brown to black and occur in the rectum, around the mouth, and on the lips, buccal mucosa, feet, nasal mucosa, and conjunctivae. These spots are typically present at puberty. [10]
The polyps most commonly appear in the small intestine (55%), followed by stomach and duodenum (30%) and the colorectal area (15%).
Although adenomas can occur concurrently in the syndrome, these polyps are mostly hamartomas of the muscularis mucosa. They appear as pedunculated lobulated lesions, measuring from a few millimeters to several centimeters. Peutz-Jeghers syndrome is inherited as an autosomal dominant trait, [19] but de novo cases can also develop. It affects all ethnic groups with equal sex distribution [10] ; however, symptoms appear earlier in males (at age 5-10 years) than in females (at age 10-15 years). [19]
GI disturbances become apparent later, usually during early adolescence. Some patients present with an increased frequency of defecation, rectal bleeding, anemia, abdominal pain, vomiting, or recurrent episodes of intussusception. [19] Prolapse of rectal polyps in the first year of life, even in the absence of pigmentation, may indicate Peutz-Jeghers syndrome, at least in the familial cases.
Compared with the general population, patients with Peutz-Jeghers syndrome have a 13-fold increased risk of death due to GI cancer and a 9-fold increased risk for all other cancers. [10] The risk of death due to cancer by age 60 years is 50%, and it reaches 85% by age 70. Adenomatous and carcinomatous changes in the hamartomas have been reported. [20]
Peutz-Jeghers syndrome is closely related to early-onset non-GI malignancies, including breast, ovary, cervix, fallopian tube, thyroid, lung, gallbladder, bile duct, pancreatic, and testicular tumors. [5]
Screening tests to detect all these forms of cancer are recommended in children who present with abdominal pain or occult anemia and melanotic-pigmented spots. An aggressive screening and biopsy program should be undertaken, including an annual examination with CBC count, breast and pelvic examinations (with cervical smears and pelvic ultrasonography) in females, mammography at age 25 years, testicular examination in males, pancreatic ultrasonography, and biennial upper and lower endoscopy.
Extensive intestinal resections are contraindicated because of the recurrent nature of the polyps and the ensuing short-bowel syndrome that may result. Rapid growth, induration, severe dysplasia, villous changes, or polyps larger than 15 mm (which present a much higher risk of malignant transformation) suggest the need for a more aggressive intervention. [10]
Gardner syndrome
In 1962, Gardner and colleagues noticed extra-colonic manifestations in some kindred with polyposis. In this syndrome, the polyps are adenomatous rather than hamartomas. [21]
The associated extraintestinal tumors include desmoid cysts, cysts of the mandible, fibromas, osteomas, and hypertrophy of the retinal pigmented epithelium. [21] Bone tumors are most common (80%), followed by inclusion cysts (35%) and desmoid tumors (18%). [12] The syndrome is inherited in an autosomal dominant pattern. Osteomas are most frequently found in the skull and facial bones. Abnormal dentition is common. [10] Periampullary malignancies may develop during the third or fourth decades of life at rates much higher than in the general population. [12]
Gardner syndrome is considered a phenotypic variant of FAP, and different mutations on the adenomatous polyposis coli (APC) gene have been shown to be associated with this syndrome (APC polymorphism in exons 13 and 15). [22] Intestinal polyps have a 100% likelihood of undergoing malignant transformation. [23]
The natural history and treatment of patients with colonic polyps are the same as in those with FAP. Desmoid tumors of the abdominal wall and mesentery occur in 20% of patients with Gardner syndrome, usually appear 6-30 months after surgery for intestinal manifestations, and are the leading cause of death in patients who have undergone colectomy. Desmoid tumors are dense fibroblastic proliferations but can present with dysplasia and even fibrosarcoma.
Treatment is challenging. When these tumors are small and well defined, excision is feasible with a recurrence rate of 10%; however, some are not identified until they become unresectable. Desmoids that involve the small bowel mesentery should be treated according to their symptoms and growth rate. Sulindac, tamoxifen, or vinblastine and methotrexate are adequate for slow-growing, mildly symptomatic tumors. Aggressive tumors require high-dose tamoxifen, or antisarcoma chemotherapy (doxorubicin and dacarbazine), and possibly radiation therapy. [24, 25]
Turcot syndrome
This syndrome, also considered a variant of FAP, includes multiple pediatric brain tumors (eg, gliomas, ependymomas) in families that also have an increased risk of polyposis and colon cancer. All patients with this syndrome develop carcinoma of the colon as young adults. [12] Colonic adenocarcinomas occur in the colonic polyps and in the mucosa between the polyps. Patients may present with chronic bloody diarrhea, hypoproteinemia, weight loss, anemia, malnutrition, bowel obstruction, and intussusception.
Hamilton et al found that families with Turcot syndrome have mutations in APC or HNPCC genes. [26] The type of brain tumor correlates with the mutations: medulloblastomas in APC -related mutations and microsatellite instability in families with glioblastoma multiforme. [10] In patients with a strong family history, begin diagnostic investigation during the second decade of life and continue annually.
Cronkhite-Canada syndrome
Cronkhite-Canada syndrome is a variant of juvenile polyposis in which the GI polyps are associated with skin hyperpigmentation, alopecia, and nail changes. Hair loss and skin and nail changes may be evident long before GI symptoms appear. The hamartomatous polyps appear in the stomach and colon. Chronic diarrhea results in malabsorption, hypovitaminosis, hypoproteinemia, and fluid and electrolyte imbalance. Because patients with Cronkhite-Canada syndrome may develop colonic malignancy, close follow-up is recommended.
Typically, as in all syndromes associated with an increased risk of cancer, it is recommended to follow a screening schedule similar to the one used for patients with Peutz-Jeghers syndrome, in order to identify malignancies at an earlier stage.
Bloom syndrome
Bloom syndrome is a rare, recessively inherited disease, in which growth retardation, accelerated aging, immunodeficiency, susceptibility to chromosome breaks, and a high frequency of malignant tumors are observed. [27] Patients with Bloom syndrome appear hypersensitive to various DNA-damaging agents, such as ultraviolet light and irradiation. A generalized DNA repair defect is present, likely a defect in DNA ligation; thus, this process has been encompassed in diseases of DNA repair defects, such as xeroderma pigmentosum, ataxia-telangiectasia, and Fanconi anemia.
The Bloom syndrome gene has been found in chromosome 15. In this regard, genes involved in DNA repair may be considered tumor suppressor genes. No specific incidence of colorectal cancer in patients with Bloom syndrome has been described in the literature.
Cowden syndrome
Cowden syndrome is an autosomal dominant transmitted disease with hamartomas of all 3 embryonal layers. Facial trichilemmomas, oral papillomas, multinodular goiter, and GI polyps with occasional GI cancer may also be found in patients with this syndrome. Fibrocystic breast disease and esophageal glycogenic acanthosis have been described. [25] These patients have a higher risk of breast and thyroid cancer. Germline mutations have been identified in the PTEN gene. Lhermitte-Duclos disease is a variant of Cowden syndrome associated with cerebellar hamartomatous overgrowth.
Treatment is directed toward alleviating symptoms of pain, bleeding, or obstruction. Polyps should be removed when symptomatic, and screening to detect subsequent development of more polyps is warranted.
Ruvalcaba-Myhre-Smith syndrome
This syndrome includes developmental abnormalities, microcephaly, and juvenile polyposis. It is a rare disease that occurs in males. No cancer has been reported in these patients. The polyps are removed when symptomatic, and family screening is advised.
Osler-Weber-Rendu syndrome
Also termed hereditary hemorrhagic telangiectasia (HHT), Osler-Weber-Rendu syndrome is an autosomal dominant familial disorder characterized by telangiectases and vascular malformations of the skin and mucous membranes and recurrent GI bleeding. It may also affect the brain, lungs, and liver. The lesions are typically noticed in the first few years of life, and 50% of patients have had GI bleeding by the age of 10 years. A family history of the disease is reported in 80% of cases. [28]
Telangiectases are usually present on the lips, oral and nasopharyngeal membranes, tongue, and perlingual areas. They also occur in the colon but are more common in the stomach and small bowel, where they tend to cause significant bleeding. [29]
In one study, 6 of 24 patients (25%) evaluated with HHT developed colonic neoplasia, 3 had adenocarcinoma of the colon, and 3 more had multiple colonic polyps. [30] Elinav et al recommend lower GI tract evaluation for all patients with new-onset anemia or GI bleeding, even if the blood loss may be a manifestation of GI HHT. [31]
Oldfield syndrome
Oldfield syndrome refers to the association between sebaceous cysts and FAP. Patients present during adolescence with subcutaneous lesions typically located on the extremities, scalp, and face; they develop during adolescence. [32] These patients share the same chromosomal derangements as those with FAP (ie, germline mutations of the APC gene on band 5q21).
Serrated polyposis syndrome
This syndrome, also known as hyperplastic polyposis syndrome, has an unknown molecular etiology and inheritance pattern, arising mostly as a sporadic syndrome; it confers a 35% risk of colorectal cancer. [33]
The colonoscopic criteria for this syndrome include one of the following:
-
At least 5 histologically confirmed serrated polyps proximal to the sigmoid colon, with at least 2 being greater than 1 cm.
-
Any number of serrated polyps proximal to the sigmoid colon, in a patient with a first-degree relative diagnosed with serrated polyposis syndrome.
-
More than 20 serrated polyps scattered throughout the entire colon.
In order to prevent malignancy in these patients, complete colonoscopic detection and resection of polyps should be performed. In cases in which this is not feasible, surgical resection is recommended. [5]
MutYH-associated polyposis syndrome
The most recently described adenomatous polyposis syndrome, MutYH-associated polyposis (MAP), has an autosomal recessive herency and requires an inherited mutation from each parent for the development of the disease. MAP is caused by a biallelic mutation in the MutYH gene, located in the chromosome 1p33-34, which encodes for a DNA glycosylase responsible for base excision repair; this mutation occurs in 1-2% of the general population. [4]
The polyps found in MAP are typically small tubular or tubulovillous adenomas. In untreated patients, the risk of colorectal cancer is 80% at age 80. In contrast to FAP, the clinical presentation is usually an adult patient with 10 to thousands of polyps, resembling a less severe FAP syndrome that presents during adulthood. [5]
Colorectal carcinoma
Syndromes associated with CRC include the following:
-
Gardner syndrome - Polyposis, osteomas, and multiple sebaceous cysts
-
Turcot syndrome - Polyposis and brain tumors (gliomas, ependymomas)
-
Peutz-Jeghers syndrome - Colonic polyposis, ovarian tumors, and mucocutaneous pigmentation of lips, oral mucosa, and perioral region
-
Cronkhite-Canada syndrome - GI polyposis, skin hyperpigmentation, alopecia, and nail changes
-
Osler-Weber-Rendu syndrome - Juvenile polyps and hepatic telangiectasia
-
Oldfield syndrome - Polyposis and multiple sebaceous cysts
-
Bloom syndrome - Growth retardation, accelerated aging, immune deficiency, and malignant tumors
-
Cowden syndrome - Hamartomas, GI polyps, breast, thyroid, and GI cancer
-
Ruvalcaba-Myhre-Smith syndrome - Microcephaly and juvenile polyposis in males; no cancer
-
Serrated polyposis syndrome
-
MutYH-associated polyposis syndrome
The screening schedule for patients with polyposis syndromes and increased risk of malignancy is as follows:
-
Assessment of symptoms related to polyps - Annually
-
Blood count to detect anemia - Annually
-
Breast and pelvic examinations with cervical smears and pelvic ultrasonography in girls - Annually
-
Testicular examination with ultrasonography in boys - Annually
-
Pancreatic ultrasonography - Annually
-
Esophagogastroduodenoscopy and colonoscopy - Biennially
-
Mammography - Recommended at ages 25, 30, 35, and 38 years; biennially until age 50 years; annually thereafter
Familial colon cancer
Most cases of CRC are not inherited as cancer-predisposition syndromes. However, hereditary colorectal cancer is not uncommon; 4-5% of cases are due to Lynch syndrome, and perhaps another 6% are caused by other various hereditary cancer syndromes. [34]
Awareness of hereditary colorectal cancer is important to family members, who can direct treatment and cancer surveillance and prevention options.
Lynch syndrome is inherited as an autosomal dominant condition, leading to increased risks of CRC, as well as endometrial, ovarian, gastric, and several other cancers. When one gene is mutated in the germline, there is a high likelihood that the individual will acquire a second somatic mutation in the other copy (allele) of that gene. Then, the cell will no longer be able to repair mismatch mutations in the DNA. [34]
Colorectal Carcinoma Genetics (Defects in Mismatch Recognition and Repair)
The unwinding and copying enzymes that replicate DNA form a highly efficient and accurate replicative complex. However, this process is not perfect. Mistakes in base pairing occasionally occur. Some stretches of DNA are more likely to accumulate errors than others, particularly stretches of DNA that consist of tandem-repeat units. These areas are termed microsatellite regions. Certain patients have marked instability in the microsatellite repeats throughout their genomes; this instability leads to a failure to recognize and repair these nucleotide mismatches. Mismatch repair defects are an early step in the process leading to malignant transformation.
The progression from normal to dysplastic colonic epithelium begins with hyperplasia, followed by the development of adenomas and, finally, invasive carcinomas. Most mutations that occur in colon cancer develop after birth in single cells as a result of exposures to environmental influences or perhaps as a result of mistakes that cells make when they copy their DNA during cell division. Approximately 80% of annual cases of colorectal carcinoma (CRC) are not associated with hereditary factors.
The progression of adenoma to carcinoma depends on reproducible genetic alterations such as APC gene inactivation, K-ras oncogene activation, and p53 mutation.
Mutations in the APC gene, a tumor suppressor gene that controls tumor initiation, are present in 80-90% of patients with familial adenomatous polyposis (FAP). [24, 35] When the APC gene is mutated, the function of both APC alleles is lost. One allele is defective at birth in all cells, having been inherited from one parent; the other APC gene allele is mutated in individual colon cells during early childhood, supporting the 2-hit hypothesis by Knudson. [36]
Malignant progression from the development of hyperplasia takes 20-30 years. This is because the tumors have to accumulate other mutations in oncogenes and other tumor suppressor genes that convert the benign adenoma into a malignant tumor.
In contrast, defects in DNA repair, particularly a DNA repair system termed DNA mismatch repair, cause hereditary nonpolyposis colon cancer (HNPCC). The enzymes that copy DNA are not perfect and often make mistakes. This mismatch must be repaired in order to avoid mutations. The DNA mismatch repair system recognizes the DNA mismatch and repairs it. [37]
Patients with HNPCC do not have defects in the APC gene inherited from their parents. Benign tumors (ie, adenomas) develop at the same rate in these patients as in the general population; however, once a patient with HNPCC has an adenoma, it rapidly progresses because of the inherited DNA repair defect. Mutations involving tumor suppressor genes and oncogenes rapidly accumulate, and, as a result, only 3-5 years are needed for a benign tumor to progress to cancer. FAP may be considered a disease of tumor initiation, whereas HNPCC may be considered a disease of tumor progression. [38]
Familial Adenomatous Polyposis
Familial adenomatous polyposis (FAP) is the most common polyposis syndrome and the second most common inherited colorectal cancer syndrome. [29, 39] It occurs in only 1 in 7000 individuals. This autosomal dominant syndrome is characterized by a mutation in the adenomatous polyposis coli (APC) gene on chromosome 5, leading to formation of numerous adenomatous polyps in the colon and rectum, beginning in the first and second decades of life. Patients with FAP have an estimated 1 in 471 risk of developing colorectal cancer before the age of 20 years, and their lifetime risk of developing the disease is nearly 100% by 40-50 years of age.
Approximately 80% of patients have a family history of FAP, while 20% appear to be de novo mutations. [40]
The APC tumor-suppressor gene encodes a large multifunctional protein product that is involved in a broad spectrum of cellular processes, such as cell cycle regulation, apoptosis, cell adhesion and migration, microtubule assembly, cell fate determination, and chromosomal stability. The main function of APC is to downregulate β-catenin, which inhibits the Wnt signaling pathway. The gene's loss of function leads to increased cellular proliferation.
FAP has classic and attenuated forms. In classic FAP, hundreds to thousands of adenomatous polyps tend to concentrate in the distal colon and rectum. Attenuated FAP is characterized by fewer (10-100) polyps, which tend to be located in the proximal large intestine. Age of onset is later than that of classic FAP, with malignant transformation also occurring 10 to 20 years later.
Cancer occurs only rarely in patients with FAP who are younger than 20 years; however, these cases are usually associated with a severe polyposis phenotype.
Extracolonic manifestations of FAP include congenital hypertrophy of the retinal pigmented epithelium; gastric and duodenal polyps; desmoid tumors and skin lesions, including fibromas, lipomas, and sebaceous cysts; dental anomalies; and osteomas of the jaw and skull. [39]
Prophylactic colectomy is the operation of choice for the management of FAP, although recommendations on the timing or age of colectomy remain unclear. Patients with classic FAP usually undergo surgery between 15 and 25 years of age. [39]
Surgical choices include the following:
-
Total proctocolectomy with ileal pouch anal anastomosis (IPAA)
-
Total abdominal colectomy with ileorectal anastomosis (IRA)
-
Proctocolectomy with ileostomy
Total proctocolectomy with IPAA is an adequate restorative procedure, which effectively minimizes the risk of colorectal cancer while maintaining continence. Abdominal colectomy with IRA has also been used; however, 30% of patients who undergo this procedure will develop rectal cancer by age 60 years. [40]
A 1-stage IPAA results in better long-term bowel control but is also associated with an increased incidence of anastomotic leakage, reoperation, and polyp recurrence and should, therefore, be reserved for selected patients. A 2-stage IPAA with diverting ileostomy seems to minimize anastomotic leaks. [39]
For colonic resection, a combination of oral and mechanical bowel preparation is associated with fewer operative complications, including anastomosis leakage, as well as superficial and intra-abdominal infections. [41]
Hereditary Nonpolyposis Colon Cancer (Lynch Syndrome)
Lynch syndrome (LS) accounts for 2-4% of all diagnosed cases of colorectal cancer (CRC). It is characterized by a relatively young age of onset (45 years) and predominantly right-sided colon cancer, but it has been identified in children aged 9-13 years. The lifetime risk of LS-associated colorectal cancer is 52-82%. [7]
Patients with LS develop polyps that advance from adenoma to carcinoma more rapidly than in sporadic cases (2-3 years versus 8-10 years) and have a lifetime risk of CRC of 70%.
Forty percent of CRC cases in patients with LS are diagnosed before age 40 years. Histologically, these colonic tumors are often mucinous, high grade, poorly differentiated, and right sided, with large numbers of tumor-infiltrating lymphocytes. [3]
DNA mismatch repair (MMR) is defective in this syndrome. MMR promotes genomic stability by correcting acid-base and small insertion/deletion loops that form during DNA replication. [7]
LS is caused by germline mutation in a mismatch DNA repair gene (human MutL homolog 1 [MLH1], human MutS homolog 2 [MSH2], human MutL homolog 6 [MSH6], human postmeiotic segregation increased 2 [PMS2], or epithelial cell adhesion molecule [EpCAM]), leading to microsatellite instability. Mutations in hMSH2 and hMLH1 account for up to 70-90% of defects identified in families with LS, mutations in MSH6 are found in 10-14% of defects, and mutations in PMS2 are found in 15% of defects. LS caused by mutations in MSH2 or MLH1 may present earlier than that caused by mutations in MSH6.
The resulting changes in sequence length, known as microsatellite instability, are a hallmark of LS. Microsatellite instability is associated with a favorable prognosis and has important implications for therapeutics in CRC.
Affected individuals often manifest café-au-lait spots, central nervous system tumors, and hematologic malignancies. Consanguinity is often noted in affected families, and parents of affected children rarely experience LS-related malignancy.
Few data on prophylactic colectomy for patients with LS are available. Carriers of a hereditary nonpolyposis colon cancer mutation rarely develop CRC in the first 2 decades of life. Prophylactic colectomy may be appropriate for patients whose colons are technically difficult to evaluate by colonoscopy, those who cannot or will not comply with screening recommendations, those with severe psychological distress resulting from fear of developing CRC, or patients with a family history of early-onset CRC. [39] MMR deficiency is not exclusive for LS-associated tumors, as it has been found in 15-20% of sporadic CRC cases. [18]
Sporadic Colorectal Carcinoma
Colorectal cancer (CRC) remains one of the top 3 carcinomas for both new diagnosis and mortality in the United States. More than a million Americans are estimated to be living with colon cancer. [42]
Epidemiology
In the general population, the lifetime risk of developing colorectal carcinoma is 5-6%. However, CRC is quite rare in children and adolescents, with a reported annual incidence of only one per million persons younger than 20 years in the United States. Only 1% of total CRC cases occur in patients younger than 30 years. [43]
Primary pediatric gastrointestinal (GI) malignancies represent fewer than 5% of all pediatric neoplasms. [43] Unfortunately, advanced stage at diagnosis, aggressive histology, and poor survival are the hallmarks of pediatric CRC. In fact, 80-90% of patients younger than 20 years present with Duke stage C/D or TNM stage III/IV. [44]
The incidence of CRC rises exponentially with increasing age from 15 to 40 years and affects both genders similarly. However, in children, the incidence of CRC is higher in males.
Although CRC rates in older adults have been dropping since 1976, the opposite trend has been observed in adolescents and young adults, in whom rates have been disproportionately higher; 18% of cases are diagnosed in persons younger than 50 years. The incidence of distal tumors has increased the most, with rectal cancer representing 44% of CRC cases in patients younger than 30 years. [1]
Risk factors
In adults, CRC is largely preventable by the adoption of a healthy lifestyle, including diet and physical activity. Obesity is a well-documented risk factor for many types of malignancies, including CRC. [45]
Diet affects the risk of developing CRC. Both red and processed meats could increase the risk, whereas fiber, fruit, and vegetables may decrease it. [46] Other dietary factors, such as fish, vitamins, minerals, and coffee, might have potential effects on the risk of developing CRC. However, factors responsible for adult CRC are unlikely to exert a major effect on children, in whom the majority of CRC cases occur spontaneously. [18]
CRC in adolescents and young adults may be associated with a familial cancer syndrome. However, whereas inherited cancer syndromes are more likely to affect a younger population, most CRC cases in adolescents and young adults are sporadic. Only 22% of adolescents and young adults with CRC have a family history of the disease. [3]
Several inherited polyposis syndromes predispose to CRC, including familial adenomatous polyposis (FAP), MutYH-associated polyposis, juvenile polyposis syndrome, Cowden syndrome, and Peutz-Jeghers syndrome. Colonoscopy with polypectomy of adenomatous polyps results in a 76-90% reduction in the incidence of colon cancer in appropriately screened individuals. Thus, for patients with inherited polyposis syndromes, colonoscopic surveillance is recommended between 10 and 15 years of age.
Proper polypectomy technique decreases the risk of residual polyps and, thus, reduces interval CRC rates. Advanced polypectomy techniques, such as endoscopic mucosal and submucosal resections, have a role in the nonsurgical management of large polyps. [47]
The timing of surgery depends on the burden of polyps, presence of dysplasia, genotype, and emotional maturity. It is often delayed until after age 18 years.
Several new syndromes have been defined that give rise to CRC. A rare form of hypermutated microsatellite-stable tumor results from mutation in the POLE gene, which plays a critical role in proofreading to identify and remove mispaired nucleotides during DNA replication. [48]
Other risk factors include prior abdominal radiation for the treatment of childhood malignancies, and inflammatory bowel disease, especially ulcerative colitis. The risk of CRC in ulcerative colitis increases with the extent of the disease and duration of inflammation. CRC risk is estimated at 2%, 8%, and 18% in the first, second, and third decades of active ulcerative colitis, respectively. [49]
By virtue of age alone, all adolescents and young adults with a new diagnosis of CRC should be referred for genetic counseling and testing.
Biology
In patients aged 21 years or younger, high-grade tumors are predominant, with signet-ring cell histology (a feature associated with advanced stage and poorer prognosis) reported in 45% of cases in one cohort. Mucinous histology was reported in 80% of adolescents and young adults and 62% of children with CRC in observational studies. [3]
Microsatellite instability among adolescents and young adults with CRC may be a favorable prognostic indicator.
CRC oncogenesis studies have shown tumors in adolescents and young adults to be more complex than those in patients 45 years and older, and often associated with P53 and PTEN mutations. Interestingly, no PIK3CA mutations have been found in patients younger than 50 years, whereas such mutations are seen in 4-7% of older patients.
Attention is currently directed toward precision medicine. Tumor and genomic mutational analyses are increasingly being used to guide treatment options for patients with malignancies. Nicholson et al studied more than 400 patients with CRC and found that loss of Bcl-2 expression is associated with decreased disease-specific and overall survival. This finding could help identify the subset of patients with a more aggressive phenotype and guide therapy choices. [50]
Particular molecular profiles may identify patients with higher-risk disease, who may benefit from more aggressive treatment or specific targeted therapies.
Pathology
Colon cancer is triggered by a series of point mutations and genetic alterations that cause normal cells to transform into adenomas that progressively become dysplastic, resulting in carcinoma foci. [51] These mutations occur in a certain sequence that determines the clinical characteristics of the tumor.
CRC arises from the mucosal surface of the bowel, generally at a site of an adenomatous overgrowth or polyp. The tumor may penetrate the bowel wall and even perforate the serosa into the omental fat, lymph nodes, liver, ovaries, and other loops of bowel. Some lesions cause bowel obstruction. Synchronous lesions may be present, with the same or different histology and stages of development. Carcinoma in situ may occur in one or more polyps. Patients with synchronous primary tumors have the same prognosis as patients with single colon cancers. [52]
The epidermal growth factor receptor (EGFR) is abnormally expressed in CRC cells (72-82%). It promotes cell division, migration, and angiogenesis and inhibits apoptosis. [53] Thus, EGFR plays an important role in the pathogenesis of CRC. Its expression is associated with poor survival and increased risk of metastasis. Italiano et al demonstrated EGFR expression in CRC metastatic cells. [54] Monoclonal antibodies and low molecular weight tyrosine kinase inhibitors may be useful in the therapeutic armamentarium for patients with CRC.
A rare "flat-type" colorectal tumor has been reported. This form tends to be more aggressive despite the small size of the tumor; it shows high-grade dysplasia and progresses rapidly to invasive cancer. Inactivation of p53 and 17p-LOH have been described in this tumor. [55] They are poorly differentiated and contain pools of mucin. Mucin-producing or signet ring adenocarcinoma is the predominant cell type; it occurs in 50% of pediatric cases compared with a 5% occurrence reported in adults. [56] These tumors may become huge. The differential diagnosis includes malignant carcinoid, leiomyosarcoma, malignant fibrous histiocytoma, and metastatic tumor from other sites.
In a series reported by La Quaglia et al, the interval from symptom onset to diagnosis was not a significant predictor of mortality; however, tumor grade was. [53] Signet ring tumors, which are more common in children and adolescents than adults, behave more aggressively and are associated with earlier penetration of the bowel wall and extension along peritoneal surfaces, which suggests more aggressive tumor biology. The mucin absorbs water, swells, and invades tissues, thus promoting spread of malignant cells. Mucin also interferes with the mucopolysaccharide-coating immune recognition of carcinoma cells. [57] Children have a worse prognosis than adult patients who are at the same disease stage. [58]
Clinical presentation
Although colorectal carcinomas constitute approximately 1% of all pediatric neoplasms, they are still the most common primary gastrointestinal malignancy. Most cases of pediatric colon cancer occur during late childhood and adolescence. [18] While the gender distribution is equal in adults, the incidence of CRC in children is higher in males, with a relative ratio of 2:1. [18, 44]
Because of its rarity, CRC is seldom suspected in children and adolescents with abdominal symptoms. Therefore, the diagnosis is often delayed. [18]
Presenting symptoms are nonspecific and include vague abdominal pain, weight loss, nausea, vomiting, anorexia, change in bowel habits (diarrhea or constipation), abdominal mass or distension, rectal bleeding, and/or intestinal obstruction. Left-sided tumors, most common among adolescents and young adults, may cause changes in stool caliber and bowel habits, whereas right-sided tumors are more likely to cause symptoms of anemia. Rectal tumors may lead to blood per rectum and tenesmus.
Advanced stage at presentation is more likely in adolescents and young adults than in older patients. The delay from symptom onset to diagnosis of CRC in adolescents and young adults often exceeds 6 months. A low suspicion rate for malignancy and a lack of screening may contribute to the delay.
Overall and event-free survival seem to be better for pediatric patients. In one study, 5-year survival rates for patients with stage 4 CRC were 18.1% for adolescents and young adults versus 6.2% for older adults. [18] Younger patients are more likely to receive systemic therapies and radiation because of sufficient functional reserve and limited comorbidity.
Clinical and laboratory investigation
In the absence of rectorrhagia or hematochezia, patients may test positive for occult blood in the stool; however, screening for fecal occult blood has not proven to be of significant value for the treatment of pediatric patients. [37] Hepatic function abnormalities may be related to metastatic involvement of the liver. Anemia is due to blood loss or malnutrition.
Although fewer than 75% of colon carcinomas in children produce carcinoembryonic antigen (CEA), levels of this protein should be determined. CEA may be a useful tool in identifying recurrent disease after resection, and an increase in CEA levels during follow-up is also related to a higher mortality rate. [12, 59] However, the role of CEA levels in the diagnosis and follow-up of CRC in children is not well established. [44]
A urine metabolites screening test for colonic adenomas is under development and may be a useful alternative to conventional fecal-based screening tests. Preliminary results show that this urine test has a sensitivity of 82.7% and a specificity of 51.2%, with a negative predictive value of 88.5%, making it a future alternative for excluding colonic polyps. [60]
A blood test that includes miRNA analysis of 5 genes implicated in CRC could constitute a new screening test. Preliminary reports show a predicted specificity of 70-95% and a predictive sensitivity of 83-91%, although more multicenter studies are required. [61]
Imaging studies
Conventional radiographic studies include barium enema with air contrast to define the tumor. Abdominal and chest computed tomography (CT) scans define spread to the liver, lungs, or enlarged lymph nodes, as well as pelvic metastases, especially to the ovaries. Colorectal cancer has the ability to metastasize through various routes, including transmural invasion and spread by continuity, intraluminal extension, and hematogenous, lymphatic, and transperitoneal routes. Kaste et al analyzed 32 patients with peritoneal metastatic implants from different primary tumors and found that 22% of these patients had colorectal cancer. [62]
CT scanning may be unable to detect intra-abdominal metastases because of lesion size, paucity of intra-abdominal fat, contiguity with the primary tumor, ascites, implant location, and adequacy of bowel opacification. Current CT scanners are able to detect implants as small as 5 mm in diameter. Magnetic resonance imaging (MRI) may further improve detection.
Colonoscopy is useful in locating the site of lesions within the large bowel. The entire length of the colon should be evaluated. Transrectal ultrasonography may help determine the extent of invasion and resectability of rectosigmoid cancer. Intraoperative ultrasonography of the liver may reveal metastases not observed in other imaging studies. [12] Radioisotope studies should include a bone scan; if the results are positive, bone marrow aspiration and biopsy are indicated to determine spread to the marrow.
Advanced imaging techniques are indicated for specific patient groups. The European Society of Gastrointestinal Endoscopy (ESGE) strongly recommends that conventional screening with white light colonoscopy in high-risk patients should be performed, as well as pancolonic conventional or virtual chromoendoscopy for patients suspected or known to have Lynch syndrome or serrated polyposis syndrome.
The ESGE also recommends that all patients with longstanding colitis undergo periodic pancolonic chromoendoscopies, with either 0.1% methylene blue or 0.1% to 0.5% indigo carmine and targeted biopsies, replacing the common practice of non-targeted 4-quadrant biopsies. [11]
Staging
In 1932, Cuthbert Esquire Dukes, the Director of the Research Laboratory at St. Mark's Hospital in London, indicated that growth of colorectal cancers followed an orderly and predictable fashion. He created a staging classification system that was later modified in 1954 by Astler and Coller. [63] The extent of the disease is determined using the modified Dukes staging scheme. According to this classification, in stage A, only the mucosa and submucosa are affected; in stage B, the disease is limited to the bowel wall; in stage C, the disease is limited to the lymph nodes; and in stage D, the patient has distant metastases, peritoneal implants, direct invasion of other viscera, or surgically unresectable tumors.
The American Joint Committee on Cancer (AJCC) published the most commonly used system to evaluate prognosis in colorectal cancer. [64] The AJCC TNM staging of colorectal cancer is described in the following table.
Table 1. American Joint Committee on Cancer TNM Staging of Colorectal Cancer (Open Table in a new window)
Primary Tumor (T) |
Nodal Involvement (N) |
Distant Metastasis (M) |
TX: Primary tumor cannot be assessed. |
Nx: Regional lymph nodes cannot be assessed. |
MX: Presence of distant metastasis cannot be assessed. |
T0: No evidence of primary tumor is present. |
N0: No evidence of regional lymph node metastases is present. |
M0: No evidence of distant metastasis is observed. |
Tis: Carcinoma in situ is present. |
N1A: Metastasis in 1 pericolic or perirectal lymph node is present. |
M1A: Distant metastasis is present in a single organ. |
T1: Tumor cells invade the submucosa |
N1B: Metastasis in 2-3 pericolic or perirectal lymph nodes is present. |
M1B: Distant metastasis is present in multiple organs. |
T2: Tumor cells invade the muscularis propria. |
N1C: Metastasis in subserosa, mesentery, or nonperitonealized pericolic or perirectal tissue without lymph node metastasis. |
|
T3: Tumor cells invade the muscularis propria into nonperitonealized pericolic or perirectal tissues. |
N2A: Metastasis in 4-6 pericolic or perirectal lymph nodes. |
|
T4A: Tumor cells perforate the visceral peritoneum. |
N2B: Metastasis in 7 or more pericolic or perirectal lymph nodes is observed. |
|
T4B: Tumor cells directly invade and adhere to other organs and structures. |
|
|
The AJCC prognostic stages are defined in the following table. [64]
Table 2. American Joint Committee on Cancer Prognostic Stages of Colorectal Cancer (Open Table in a new window)
Stage |
Primary Tumor (T) |
Nodal Involvement (N) |
Distant Metastasis (M) |
0 |
Tis |
N0 |
M0 |
I |
T1 |
N0 |
M0 |
|
T2 |
N0 |
M0 |
IIA |
T3 |
N0 |
M0 |
IIB |
T4A |
N0 |
M0 |
IIC |
T4B |
N0 |
M0 |
IIIA |
T1/T2 |
N1A/N1B/N1C |
M0 |
|
T1 |
N2A |
M0 |
IIIB |
T3/T4A |
N1A/N1B/N1C |
M0 |
|
T2/T3 |
N2A |
M0 |
|
T1/T2 |
N2B |
M0 |
IIIC |
T4A |
N2A |
M0 |
|
T3/T4A |
N2B |
M0 |
|
T4B |
N1A/N1B/N1C/N2A/N2B |
M0 |
IVA |
Any T |
Any N |
M1A |
IVB |
Any T |
Any N |
M1B |
Treatment
Surgical resection remains the mainstay of treatment for colorectal carcinoma in adult and adolescent patients, and the sole therapy required for patients with stage I and II disease. [42]
Open colectomy has been the standard of care for the past 100 years. Complete en bloc tumor resection, including the lymphatic basin of the affected segment, has the greatest impact on survival. [18]
Treatment guidelines are based largely on evidence from older adults. Multidisciplinary care is crucial, and prompt referral to centers experienced in the care of adolescents and young adults should be considered for young patients.
Surgical site infection complicates approximately 15% of colectomy procedures. Oral antibiotic bowel preparation before elective colorectal surgery is associated with shorter postoperative length of stay and lower 30-day readmission rates, primarily because of fewer readmissions for infections. [47] Moghadamyeghaneh et al demonstrated that a combination of mechanical and oral antibiotic preparations significantly decreased postoperative morbidity. [41]
Enhanced recovery protocols, which aim to streamline and standardize perioperative care, have demonstrated efficacy in reducing length of stay but have not resulted in reduced readmission rates. These protocols avoid the use of bowel preparation and epidurals, and encourage early ambulation, early feeding, and early transition to oral analgesia.
Restorative proctocolectomy with ileal pouch anal anastomosis (IPAA) is a viable procedure in pediatric patients, with acceptable morbidity and good long-term results with regard to gastrointestinal function, quality of life, and patient satisfaction. [65, 66] Concerning the type of anastomosis, these authors favor stapled IPAA for prophylactic colectomy, reserving hand-sewn IPAA for patients with neoplasia. The latter is a prudent approach, because earlier dysplasia and colorectal neoplasia are the most important risk factors for developing pouch-related cancer. [65]
An ileal J-pouch is easy to create and delivers acceptable outcomes. However, an S-pouch provides an extra 1-2 cm of length, allowing for tension-free IPAA. [66]
Because children have a long life expectancy, both functional outcomes and control of neoplastic activity of the anal canal and ileal reservoir are of utmost importance. [66]
Best results are obtained with stapled, tension-free anastomosis. Intact tissue rings, good hemostasis, and absence of air leak are imperative. Malnutrition (albumin level, < 3.5 g/dL), neoadjuvant drug toxicity, anemia (hemoglobin level, < 13.5 g/dL), and prolonged high-dose corticosteroid therapy (20 mg of prednisone daily for longer than 3 months) are prognostic factors for perioperative morbidity. Perioperative complications are not uncommon (10%); they include pelvic sepsis, anastomotic leak or stricture, pouchitis, pouch failure, bowel obstruction, and anastomotic stricture. [66]
Laparoscopic resection offers similar postoperative outcomes, pouch function, and long-term quality of life compared with open procedures. [66] However, the learning curve for laparoscopic colectomy remains steep. The need to retract multiple organs, identify complex anatomy, and control large vessels makes the operation difficult. [42]
Patient selection in minimally invasive surgery for colon cancer
Good surgical outcomes depend on careful patient selection. Historic contraindications to a minimally invasive approach include elderly or high-risk patients, multiple previous abdominal operations, technically challenging surgery, and patients with serious comorbidities. All these factors can add a level of difficulty to the performance of an operation, and they are certainly associated with increased morbidity and mortality. But clearly none of these factors are hard but rather are relative contraindications to minimally invasive surgery. The surgeon’s main goal is the performance of a safe and adequate operation. [42]
Laparoscopic surgery has been found to be equal to open surgery in terms of oncologic outcome, margins, lymph node sampling, recurrence, and disease-free survival. The ability to obtain an R0 resection with tumor-free margins is a keystone of oncologic colectomy. [42]
As of now, the following are the only 2 absolute contraindications to laparoscopic CRC surgery [42] :
-
A tumor large enough that the extraction site to remove it would be of sufficient size to perform the entire operation.
-
A high-grade large bowel obstruction leading to a reduced intra-abdominal domain. The impaired visibility coupled with the friability of the tissue associated with a cancer makes minimally invasive surgery an ill-advised approach in this setting.
Advantages of minimally invasive surgery in colon cancer
In adults, benefits of minimally invasive surgery include decreased length of stay, smaller incisions, less narcotic use, less blood loss and lower transfusion rates, and improved pulmonary function after surgery. No long-term differences have been demonstrated regarding oncologic outcomes. [42]
Surgical colon cancer guidelines widely cite consensus for a goal of at least 12 lymph nodes for an adequate staging. There is a positive correlation between the number of lymph nodes examined and survival for patients with stage II and III CRC. No difference has been shown between open and laparoscopic approaches regarding the number of lymph nodes examined, as well as local or distant recurrence rates. [42]
Early reports of a high rate (21%) of port site recurrence raised concerns about minimally invasive surgery. However, larger multicenter, randomized, controlled trials have not shown any significant increase in wound site recurrence. Both laparoscopic and open CRC resections have wound recurrence rates of less than 1%. [42]
Treatment of early-stage disease (non-metastatic)
Radical surgery is the pillar of curative treatment, including en bloc resection of adjacent organs infiltrated by the tumor. A margin of 5 cm of bowel both proximal and distal to the tumor should be removed (although this distal margin may not be feasible with a low-lying rectal tumor). Right hemicolectomy is indicated for tumors proximal to the splenic flexure, whereas left hemicolectomy is carried out for tumors of the descending colon.
Rectal cancer surgery should include total meso-rectal excision (TME) to minimize the risk of local recurrence. It also requires retrieving a minimum number of lymph nodes. The utility of this approach is still uncertain for adolescents and young adults.
Radical resection of low-lying rectal cancers has traditionally required abdominoperineal resection that involves permanent colostomy, which can have significant impact on the quality of life of adolescents and young adults. For very superficial tumors, mucosal resections might be adequate. Trans-anal TME allows for proper oncologic resection without permanent colostomy.
Indications should be made in the context of an integrated treatment strategy established by an interdisciplinary local or reference board. [67]
Treatment of metastatic disease
Metastatic disease occurs in half of patients with CRC. Metastasis can spread through hematogenous, lymphatic, transcoelomic, endoluminal, or contiguity routes, and it may occur in lymph nodes, liver, lung, peritoneum, brain, and bone. Synchronous presentation at diagnosis occurs in 21% of patients and is associated with worse survival than metachronous disease.
Although most cases of metastatic CRC are incurable, combination chemotherapy with improved surgical techniques and radiation therapy plays a role in prolonging survival and reducing cancer-related symptoms in the context of extensive metastatic disease. [2]
The staging work-up for metastatic disease includes CT of the chest, abdomen, and pelvis and consideration of positron emission tomography (PET)/CT for selected patients with indeterminate findings. Routine laboratory work-up, measurement of carcinoembryonic antigen (CEA) level, and determination of RAS and BRAF status at the primary tumor should be performed. Biopsy of metastases may be useful in the setting of first recurrence or in the case of indeterminate imaging findings.
A multidisciplinary evaluation is of critical importance for most patients with metastatic CRC, because the choice and order of therapies differ depending on presentation, number of sites and location of metastases, and potential for surgical resection. [2]
Research is focused on antiangiogenic agents, antibodies against EGFR (eg, cetuximab), and sequential lines of therapy. Median survival for patients with stage IV disease now exceeds 2.5 years.
Immune checkpoint inhibitors are a novel class of antineoplastic drugs that promote T-cell activation to overcome immune evasion of cancer cells. Other novel agents include monoclonal antibodies to block the programmed cell death protein. This class of therapeutics has shown promising activity for CRC with microsatellite instability, but not for microsatellite stable tumors.
Patients with type 2 diabetes who are being treated with metformin and in whom CRC develops have lower morbidity and better tumor-free survival than patients who are not receiving metformin. [68] .
Tumor debulking offers little survival benefit for patients with extensive metastatic disease. [44] However, in the case of a limited burden of cancer spread, surgical resection of metastatic deposits may result in a longer disease-free interval or even a cure in a minority of cases. [3]
Metastasis limited to the liver, lung, or peritoneum may be operable. Resection of peritoneal disease is often performed in conjunction with heated intraperitoneal chemotherapy (HIPEC), although the added value of this therapy compared with surgery alone remains uncertain.
Chemotherapy and radiation therapy
Radiation is often given with or without chemotherapy in the neoadjuvant setting for rectal cancer, to reduce the risk of local recurrence.
Adjuvant chemotherapy may be considered for recurrence risk reduction in stage III or high-risk stage II CRC. Features that indicate a high risk of recurrence in stage II CRC include the following:
-
Peritoneal tumor infiltration (T4)
-
High grade
-
Perforation or obstruction at presentation
-
Lymphovascular or perineural invasion
-
Inadequate lymph node sampling
Combination chemotherapy with a fluoropyrimidine and oxaliplatin for 6 months remains the standard of care, although studies show little difference between 3 and 6 months of therapy in the absence of T4 or heavy lymph node involvement.
After surgery and adjuvant therapy, surveillance monitoring for recurrence includes periodic clinical assessment, CT, colonoscopy, and measurement of CEA levels.
Prognosis
Colorectal cancer during childhood is associated with a poor prognosis, mainly attributed to delayed diagnosis, greater tumor virulence, and the advanced stage of disease at presentation. Up to 50% of CRC cases that present during childhood are mucinous adenocarcinomas, and 60% have evidence of metastatic disease at presentation. [18]
Impact of treatment on fertility
Adhesions following pelvic surgery contribute to infertility risk in women. Pelvic radiation for rectal cancer is likely to cause premature ovarian failure. Options to prevent this complication include pre-radiation laparoscopic ovarian transposition or cryopreservation.
Chemotherapy can also impair reproductive health. Fluorouracil is not likely to cause infertility, but oxaliplatin has been associated with moderate infertility risk in animal studies.
Genetic testing and chemopreventive agents
Genetic testing for cancer susceptibility has positive attributes. Family members who test negative for the particular APC or MMR gene are spared the repeated medical and endoscopic examinations that otherwise would have to be performed. They are also spared the anxiety associated with not knowing whether they are affected. Surveillance can then be concentrated on those who have inherited a mutant gene.
Although genetic testing studies for screening are lacking, expert opinion recommends that genetic testing should be performed in every patient with newly diagnosed CRC, to prevent morbidity and mortality in direct relatives. Implementation of universal screening for Lynch syndrome has been declared as part of the 2020 objectives of the Office of Public Health Genomics in the United States. [15]
Future prospective studies should include a systematic family history and testing for potentially relevant genetic conditions if indicated. Trials should also address the risk of CRC in family members and promote sensible family screening guidelines. [69]
Most cancers have an increased glycolytic pathway (Warburg effect). Proteomic studies have demonstrated this pathway enhancement in CRC culture cells. Bi et al also reported on decreased gluconeogenesis, suppressed glucuronic acid pathway, and an impaired tricarboxylic acid cycle. [70] Their findings provide an insight into the tumorigenesis of CRC and possible specific targeted therapies to be developed.
Chemopreventive agents may one day inhibit the development of adenomas. Nonsteroidal anti-inflammatory drugs (NSAIDs) have a well-documented effect on shrinking existing adenomas in patients with familial adenomatous polyposis and potentially inhibit their formation. [36]
Although NSAIDs may have non–cyclooxygenase (COX)-mediated pathways, aspirin inhibits COX enzymes in the conversion of arachidonic acid to prostaglandins. COX-1 is thought to produce cytoprotective prostaglandins in the GI tract, whereas COX-2 is expressed in response to growth factors, mitogens, and cytokines and is found in 50% of colorectal adenomas and in 85% of cancers. [25] COX-2–specific inhibition is believed to be protective against epithelial transformation. Nuclear factor kappa B (NF-κB) transcription factor translocation that produces apoptosis is another mechanism of aspirin.
Specific COX-2 inhibitors are no longer used because of their cardiovascular adverse effects. Bevacizumab is a monoclonal antibody that targets vascular endothelial growth factor-A (VEGF-A), which is believed to be critical in cancer angiogenesis. Cetuximab is another monoclonal antibody that targets the EGFR, which is involved in cancer cell proliferation, degradation of the extracellular matrix (invasiveness), tumor migration, and endothelial proliferation. It may well become a good genetic target treatment for CRC. Collaborative multi-institutional pediatric clinical trials are needed to evaluate the prognosis, optimal treatment response, and basic biology of childhood-onset CRC.
Questions & Answers
Overview
What are pediatric colorectal tumors?
What is the prevalence of pediatric colorectal tumors?
What is Peutz-Jeghers syndrome?
What is Cronkhite-Canada syndrome?
What are colonic polyposis syndromes?
How are juvenile polyposis syndromes classified?
What are lymphoid polyps (lymphoid nodular hyperplasia)?
What is juvenile polyposis syndrome (JPS)?
What is diffuse juvenile polyposis of infancy?
What is diffuse juvenile polyposis?
What is juvenile polyposis coli?
What is Ruvalcaba-Myhre-Smith syndrome?
What is Osler-Weber-Rendu syndrome?
What is serrated polyposis (hyperplastic polyposis syndrome)?
What is MutYH-associated polyposis (MAP)?
Which polyposis syndromes are associated with colorectal cancer?
What is included in the long-term monitoring of patients with polyposis syndromes?
What is the prevalence of hereditary colorectal cancer?
What is the role of genetics in the pathogenesis of colorectal tumors?
What is familial adenomatous polyposis (FAP)?
How common are colorectal tumors?
What is the prevalence of colorectal cancer (CRC)?
What are the risk factors for colorectal cancer (CRC)?
What is the biology of pediatric colorectal tumors?
What is the pathophysiology of pediatric colorectal tumors?
Which clinical history findings are characteristic of pediatric colorectal tumors?
What are the reported survival rates for pediatric colorectal tumors?
What is the role of lab tests in the workup for pediatric colorectal tumors?
What is the role of imaging studies in the workup of pediatric colorectal tumors?
How are pediatric colorectal tumors staged?
How are pediatric colorectal tumors treated?
When is laparoscopic surgery contraindicated for the treatment of colorectal tumors?
What are the benefits of laparoscopic surgery for the treatment of colorectal tumors?
How are early-stage pediatric colorectal tumors treated?
How are metastatic pediatric colorectal tumors treated?
What is the prognosis for pediatric colorectal tumors?
What is the effect on fertility of pediatric colorectal tumor treatments?
What is the role of genetic testing in the prevention of pediatric colorectal tumors?
Which medications may have a role in the prevention of pediatric colorectal tumors?
-
This picture depicts an abdominal CT scan of a 7 year-old boy with a mucinous adenocarcinoma of the ascending colon. Note the thickness and increased vascularity of the colonic wall, as well as irregularities on the serosal surface. This cut also shows severe tumor infiltration of the colonic mesentery surrounding the mesenteric and retroperitoneal vessels.
-
Coronal CT scan demonstrating the profuse tumoral infiltration of the ascending colonic mesentery surrounding mesenteric and portal vessels. Also note the thickness of the colonic hepatic flexure.
-
Surgical specimen after right hemicolectomy, including the terminal ileum up to the transverse colon. Mesenteric fat, vessels and lymph nodes were resected en block with the ascending colon. The large intestine has been opened longitudinally. Note the tumor on the right lower quadrant of the image, with severe thickness of the wall, areas of necrosis and hemorrhage, and some stippled calcifications.
Tables
Primary Tumor (T) |
Nodal Involvement (N) |
Distant Metastasis (M) |
TX: Primary tumor cannot be assessed. |
Nx: Regional lymph nodes cannot be assessed. |
MX: Presence of distant metastasis cannot be assessed. |
T0: No evidence of primary tumor is present. |
N0: No evidence of regional lymph node metastases is present. |
M0: No evidence of distant metastasis is observed. |
Tis: Carcinoma in situ is present. |
N1A: Metastasis in 1 pericolic or perirectal lymph node is present. |
M1A: Distant metastasis is present in a single organ. |
T1: Tumor cells invade the submucosa |
N1B: Metastasis in 2-3 pericolic or perirectal lymph nodes is present. |
M1B: Distant metastasis is present in multiple organs. |
T2: Tumor cells invade the muscularis propria. |
N1C: Metastasis in subserosa, mesentery, or nonperitonealized pericolic or perirectal tissue without lymph node metastasis. |
|
T3: Tumor cells invade the muscularis propria into nonperitonealized pericolic or perirectal tissues. |
N2A: Metastasis in 4-6 pericolic or perirectal lymph nodes. |
|
T4A: Tumor cells perforate the visceral peritoneum. |
N2B: Metastasis in 7 or more pericolic or perirectal lymph nodes is observed. |
|
T4B: Tumor cells directly invade and adhere to other organs and structures. |
|
|
Stage |
Primary Tumor (T) |
Nodal Involvement (N) |
Distant Metastasis (M) |
0 |
Tis |
N0 |
M0 |
I |
T1 |
N0 |
M0 |
|
T2 |
N0 |
M0 |
IIA |
T3 |
N0 |
M0 |
IIB |
T4A |
N0 |
M0 |
IIC |
T4B |
N0 |
M0 |
IIIA |
T1/T2 |
N1A/N1B/N1C |
M0 |
|
T1 |
N2A |
M0 |
IIIB |
T3/T4A |
N1A/N1B/N1C |
M0 |
|
T2/T3 |
N2A |
M0 |
|
T1/T2 |
N2B |
M0 |
IIIC |
T4A |
N2A |
M0 |
|
T3/T4A |
N2B |
M0 |
|
T4B |
N1A/N1B/N1C/N2A/N2B |
M0 |
IVA |
Any T |
Any N |
M1A |
IVB |
Any T |
Any N |
M1B |

What would you like to print?
- Practice Essentials
- Background
- Polypoid Disease of the Gastrointestinal Tract
- Colorectal Carcinoma Genetics (Defects in Mismatch Recognition and Repair)
- Familial Adenomatous Polyposis
- Hereditary Nonpolyposis Colon Cancer (Lynch Syndrome)
- Sporadic Colorectal Carcinoma
- Questions & Answers
- Show All
- Media Gallery
- Tables
- References






