Practice Essentials
Malignant rhabdoid tumor (MRT) is one of the most aggressive and lethal malignancies in pediatric oncology. (See the image below.) Although mutations or deletions of the SMARCB1 gene play a role in the development of MRT, the events that incite these genetic alterations are unknown. Several cases of familial MRT are reported. No environmental or infectious associations with MRT have been established.
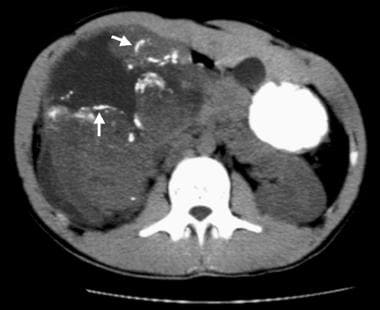 Nonenhanced CT scan demonstrates linear and curvilinear calcifications outlining tumor lobules in a malignant rhabdoid tumor (MRT) (arrows). A hypoattenuating fluid collection surrounds and separates the lobules. These imaging features are seen with MRT more often than with other childhood renal neoplasms.
Nonenhanced CT scan demonstrates linear and curvilinear calcifications outlining tumor lobules in a malignant rhabdoid tumor (MRT) (arrows). A hypoattenuating fluid collection surrounds and separates the lobules. These imaging features are seen with MRT more often than with other childhood renal neoplasms.
Signs and symptoms
Children with MRT of the kidney present with signs and symptoms related to an intrarenal mass. Pain is difficult to assess because the median age at presentation is about 11 months. However, fussiness is reported in most patients.
Gross hematuria is a presenting feature in about 60% of patients, and fever is a presenting symptom in 50% of patients with an MRT of the kidney. Up to 20% of patients have synchronous or metachronous CNS lesions.
See Presentation for more detail.
Diagnosis
MRT is definitively diagnosed by means of histologic analysis.
Laboratory studies
The following tests may be helpful in the workup:
-
Complete blood cell (CBC) count: Approximately 55% of patients with MRT present with a hemoglobin level of less than 9 g/dL.
-
Urinalysis: Microscopic hematuria is seen in 75% of patients with MRT.
-
Serum calcium measurement: Up to 25% of patients with MRT present with hypercalcemia.
Imaging studies
The following imaging studies are suggested for the diagnosis and staging of MRT:
-
Abdominal computed tomography (CT) or magnetic resonance imaging (MRI)
-
Chest CT
-
Abdominal ultrasonography
-
MR, CT, fluorodeoxyglucose (FDG), and/or positron emission tomography (PET) imaging of the brain
-
Bone scanning (under debate if this is necessary)
See Workup for more detail.
Management
After the primary tumor is surgically removed, chemotherapy and radiation are indicated as adjuvant therapy. The treatment for MRT remains investigational. No accepted standard therapy has been established for this disease. Enrollment of patients in clinical trials is strongly encouraged.
See Treatment and Medication for more detail.
Background
Introduction
Malignant rhabdoid tumor (MRT) was initially described in 1978 as a rhabdomyosarcomatoid variant of a Wilms tumor because of its occurrence in the kidney and because of the resemblance of its cells to rhabdomyoblasts. The absence of muscular differentiation led Haas and colleagues to coin the term rhabdoid tumor of the kidney in 1981. [1]
Although renal malignant rhabdoid tumor was historically included in treatment protocols of the National Wilms Tumor Study (NWTS) Group, this tumor is now recognized as an entity separate from a Wilms tumor. In contrast to a Wilms tumor, a MRT of the kidney is characterized by the early onset of local and distant metastases and resistance to chemotherapy. Whereas the overall survival rate for Wilms tumors exceeds 85%, the survival rate for renal MRTs is only 20-25%.
Since MRT of the kidney was originally described, MRTs have been reported in practically every location in the body, including the brain, liver, soft tissues, lung, skin, and heart. This article focuses on renal and extrarenal MRTs that arise outside the CNS.
Molecular genetics
Cytogenetic, fluorescence in situ hybridization (FISH), and loss-of-heterozygosity (LOH) studies have revealed that MRTs frequently contain deletions at chromosome locus 22q11.1. Positional cloning efforts revealed that this locus contains the SWI/SNF related, matrix-associated, actin-dependent regulator of chromatin, subfamily B, member 1 (SMARCB1) gene, also known as human sucrose non-fermenting gene number 5 (hSNF5), integrase interactor 1 (INI1), or 47-Kd Brg1/Bam-associated factor (BAF47). [2] SMARCB1 encodes a member of the human SWI/SNF complex.
Combined analyses including FISH, coding sequence analysis, high-density single nucleotide polymorphism-based oligonucleotide arrays, and multiplex ligation-dependent probe amplification enable the identification of biallelic, inactivating perturbations of SMARCB1 in nearly all MRTs, consistent with the 2-hit model of tumor formation. [3] Thus, SMARCB1 is presumed to function as a classic tumor suppressor and the primary gene responsible for MRT development.
Homozygous inactivation of SMARCB1 in mice demonstrates embryonic lethality, whereas heterozygous SMARCB1 mice demonstrate a normal phenotype at birth, with 20% developing sarcomas at a median age of 1 year. Similar to human MRTs, murine tumors in these mice acquire a second hit to the SMARCB1 locus. All mice harboring a conditional biallelic inactivation of SMARCB1 develop cancer with a median onset of 11 weeks, revealing one of the most aggressive cancer predisposition genotype-phenotype correlations known.
Unexpectedly, despite an aggressive clinical pattern of behavior, MRTs are generally diploid and genomically stable, without recurrent gene amplifications or deletions. Therefore, the mechanism by which SMARCB1 perturbation leads to aggressive neoplasia likely relates to its role in epigenetic modification. The SWI/SNF complex acts in an adenosine triphosphate (ATP)–dependent manner to remodel chromatin, which regulates gene transcription and DNA repair. Whole-exome sequencing studies of primary MRTs have shown that biallelic mutations or copy number alterations of SMARCB1 seem to be both necessary and sufficient to cause cancer. [4]
The distribution of SMARCB1 and chromosome 22-inactiving mutations, deletions, and copy number loss of heterozygosity (CN-LOH) in 200 sporadic AT/RTs, renal MRTs, and extrarenal MRTs is shown in Table 1 below. In the majority of tumors (43%), there is a mutation in one allele, and the second copy of the gene is lost as a result of a structural deletion in 22q11.2, monosomy 22, or an acquired CN-LOH event. Compound heterozygous mutations are infrequent in these patients (4%). Partial deletions and duplications are detected in approximately 15% of tumors. Homozygous deletions of exons 1-9 of SMARCB1 are present in approximately 40% of rhabdoid tumors overall, although there is an unequal distribution with respect to anatomic location. Approximately 25% of AT/RTs, 40% of renal MRTs, and 70% of extrarenal MRTs have homozygous deletions of the entire locus. [4]
Table 1. Acquired SMARCB1 Alterations in 200 Sporadic Rhabdoid Tumors (Adapted from Geller JI, Roth JJ, Biegel JA. Biology and Treatment of Rhabdoid Tumor. Crit Rev Oncog. 2015. 20 [3-4]:199-216) (Open Table in a new window)
Allele 1 Alteration |
Allele 2 Alteration |
Total |
|||
Mutation |
Partial Gene Deletion/Duplication |
Whole Gene Deletion |
CN-LOH |
||
Mutation |
8 (4%) |
1 (0.50%) |
58 (29%) |
27 (13.5%) |
94 (47%) |
Partial Gene Deletion/Duplication |
- |
5 (2.5%) |
14 (7%) |
11 (5.3%) |
30 (15%) |
Whole Gene Deletion |
- |
|
76 (38%) |
- |
76 (38%) |
Total |
8 (4%) |
6 (3%) |
148 (74%) |
38 (19%) |
200 (100%) |
Although SMARCB1 is the predominant gene altered in MRTs, approximately 2-3% retain expression of the SMARCB1 protein on immunohistochemistry and do not display inactivating mutations in the gene. [4] A small number of families and patients with MRT have been shown to have germline or somatic mutations in SMARCA4, which is the primary ATPase in the SWI/SNF complex. [5, 6]
Pathophysiology
The histogenetic origin of malignant rhabdoid tumor (MRT) remains obscure. Rhabdoid tumor cells are polyphenotypic, with an immunostaining pattern that shows evidence of mesenchymal, epithelial, and neural differentiation. Polyantigenic expression suggests that MRTs arise from a pluripotent cell capable of differentiating along several lines.
Considerable debate has been focused on whether extrarenal MRTs are the same as renal MRTs. The recognition that CNS AT/RTs have deletions of the SMARCB1 gene indicates that MRTs of the kidney and brain are closely related entities. This observation is not surprising because rhabdoid tumors at both locations possess similar histologic, clinical, and demographic features. Moreover, 10-15% of patients with non-CNS MRTs have synchronous or metachronous brain tumors, many of which are second primary AT/RTs.
More convincingly, the same germline mutations (outside of frameshift mutations) predispose carriers to AT/RT, renal MRTs, and to a lesser extent extrarenal MRTs. The majority of extrarenal MRTs are sporadic and arise as a consequence of homozygous loss of SMARCB1. The most frequent second hit in patients with a germline mutation is a large 22q deletion or monosomy 22, or a copy number loss of heterozygosity (CN-LOH) generating event. [4]
Reports to date have demonstrated that SMARCB1 loss can promote cell cycle progression resulting from upregulation of targets of the p16INK4a-Rb-E2F pathway, primarily including CyclinD1 as well as several cyclin-dependent kinases (CDK). Rb family loss has been shown to increase MRT tumorigenesis and progression, whereas ablation of CyclinD1 abrogates MRT evolution in mouse models. Similarly, tumor development in SMARCB1-deficient mice is greatly accelerated in the absence of functional p53 protein. These findings suggest a cooperative effect between SMARCB1 and the pRB, CyclinD1, and Tp53 pathways. The loss of SMARCB1 is postulated to result in a global failure of the repressive H3K27 trimethylation mark present on bivalently modified histones, mediated by the polycomb complex 2, resulting in widespread epigenetic modifications and leading to arrested development and abnormal proliferation. Two members of the polycomb complex 2, CBX6 and EZH2, are upregulated in MRT. [7] Aurora-A-kinase is also expressed in high levels in MRT and is repressible with SMARCB1 reintroduction into rhabdoid tumor cells via transcriptional down-regulation. SMARCB1 loss also leads to increased expression of GLI1, supporting a role in the biology of the sonic hedgehog pathway. [8] Bromodomain containing protein 9 (BRD9) is also a subunit of the SWI/SNF complex that is involved in epigenetic mechanisms such as regulation of transcription, chromatin remodeling and histone modification. [9] These findings suggest possible therapeutic targets for MRT.
For CNS MRT (AT/RT), biologic heterogeneity has been discovered. There are three distinct molecular subgroups of AT/RT tumors, termed ATRT-TYR, ARTR-SHH, and ATRT-MYC, that are associated with differences in demographics, tumor location, and type of SMARCB1 mutations. [10] The ATRT-TRY group is composed of mostly infratentorial tumors with broad SMARCB1 deletions and overexpression of melanosomal genes. The ATRT-SHH group is associated with both supra- and infratentorial lesions, has focal SMARCB1 aberrations, and shows overexpression of SHH pathways. The ATRT-MYC group is comprised of mostly supratentorial tumors with focal SMARCB1 deletions and over-expression of the MYC and HOX cluster. [10] The development of these molecular subgroups with associated regulatory networks and pathways will help to develop more effective, subgroup specific treatment options.
Similarly, comprehensive genomic analysis was performed on non-CNS MRTs through the Therapeutically Applicable Research to Generate Effective Targets (TARGET) initiative. [11] This analysis showed evidence for epigenetic reprogramming of HOX genes with loss of H3K27me3 marks, as well as dysregulated expression of genes involved in neural crest development and of oncogenes and tumor suppressor genes. Although SMARCB1 alterations were shown to be the dominant genetic driver in MRTs, the effects of that loss on transcriptional regulation were not uniform across all cases. Conversely, the spectrum of tumors characterized by mutations in the SMARCB1 gene has also been expanded beyond tumors with a rhabdoid histologic phenotype to include hereditary schwannomas, extraskeletal myxoid chondrosarcoma, [12] proximal-type epithelioid sarcoma, epithelioid malignant peripheral nerve sheath tumor, renal medullary carcinoma, [13] chordoma, and pediatric undifferentiated sarcoma lacking rhabdoid features. [14] Inactivation of SMARCB1 has also been identified in the small cell undifferentiated variant of hepatoblastoma, which may suggest that such tumors are better characterized as hepatic rhabdoid tumors. [15, 16] Whether extrarenal MRTs have the same histogenetic origin as that of their renal counterparts is unclear. Although some extrarenal MRTs are considered to be undifferentiated sarcomas or carcinomas with rhabdoid features, others represent true rhabdoid tumors because they have documented SMARCB1 mutations. [17] To date, the only tumor outside of MRT to demonstrate biallelic inactivation of SMARCA4, consistent with a cancer-predisposing germline mutation and second somatic alteration, is small cell carcinoma of the ovary, hypercalcemic type (SCCOHT). [18] Based on the early age at presentation and presence of rhabdoid-appearing cells on histology, it has been proposed that SCCOHT represents another type of extrarenal MRT. [19]
The Children's Oncology Group (COG) has initiated an effort to prospectively screen all types of MRT for SMARCB1 mutations and protein expression, which should improve the classification and prognostication of tumors with rhabdoid features. As molecular-based targeted therapies emerge, the distinction between true and pseudorhabdoid tumors may prove to have important therapeutic implications.
For details about the gross and histologic features of MRTs, see Histologic Findings.
Epidemiology
United States statistics
Malignant rhabdoid tumor (MRT) is a rare tumor. According to registration data from NWTS 1-5, MRT of the kidney accounts for only 158 (1.6%) of 10,031 registrants with childhood renal tumors. Likewise, only 26 (0.9%) of 3000 participants in the Intergroup Rhabdomyosarcoma Studies I-III had tumors consistent with MRT. Of the first 4,000 patients enrolled on the COG study AREN03B2, renal and extrarenal MRTs accounted for 3.71% combined. [20] About 15 cases of extrarenal, non-CNS MRTs are diagnosed each year in the North America. MRTs in infants (age 0-12 months) account for 18% of renal tumors, 14% of soft tissue tumors, and 9% of liver tumors. [21]
International statistics
The incidence of MRT in most countries has not been reported. Between 1984 and 1999, approximately 6 patients per year diagnosed with MRT were enrolled onto various national registries or protocols in Germany. [22] Between 1993 and 2005, a total of 207 renal MRT patients were enrolled on SIOP renal tumor treatment study group protocols. [23] Within the UK and Germany the age standardized annual incidence rates of extracranial MRT are 5-5.7 per million at age 1 and decrease to 0.1-0.2 at age 5. [24]
Race-, sex-, and age-related demographics
MRT has no apparent racial predilection.
MRT occurs slightly more frequently in male individuals than in female individuals, with male-to-female ratio of 1.37:1. [25]
The median age at presentation is 10.6 months, with a mean age of 15 months. Most patients are younger than 2 years. MRT has been reported in children older than this and in adults, but whether older patients have a biologically distinct subtype of MRT is unclear.
Prognosis
The prognosis for children with MRTs remains fair to poor, depending on the stage of the tumor at presentation, the patient's age at diagnosis, and possibly the genetic background. The hope is that new multi-institutional clinical trials will help in identifying novel therapies that improve the outcome of patients with this disease.
Morbidity/mortality
The overall survival rate for patients with MRT enrolled in NWTS 1-5 was 23.2%. [25]
MRT is a rapidly progressive tumor, with most deaths occurring within 12 months of presentation. The most common sites of metastasis at presentation are the lungs, abdominal lymph nodes, liver, brain, and less commonly bone (1.4%).
A young age at diagnosis is strongly associated with an adverse outcome. Four-year event-free survival rates according to age at diagnosis were 8.8% for patients aged 0-5 months, 17.2% for patients aged 6-11 months, 28.6% for patients aged 12-23 months, and 41.1% for patients aged 24 months or older (p < 0.0001). [25]
High-stage (Stage III/IV) disease is also correlated with an adverse outcome (p=0.014); most patients present with Stage III or IV disease.
The overall survival of patients with MRT in NWTS was as follows:
-
Stage I - 15 patients (33.3%)
-
Stage II - 25 patients (46.9%)
-
Stage III - 58 patients (21.8%)
-
Stage IV - 41 patients (8.4%)
-
Stage V - 3 patients (0%)
A recent study of 100 patients with extracranial MRT recruited within EU-RHAB (2009-2018) showed a 5-year overall survival (OS) rate of 45.8 ± 5.4%. [24] In univariate analyses, age at diagnosis (≥12 months), localized disease, absence of synchronous tumors, absence of a SMARCB1 germline mutation, gross total resection, radiotherapy, and achieving a complete response (CR) were significantly associated with favorable outcomes. In an adjusted multivariate model, the presence of a SMARCB1 germline mutation, distant metastatic disease, and lack of a gross total resection were the strongest significant negative predictors of outcome.
Of 53 extracranial MRT patients treated at Bejing Children's Hospital between 2007-2017, 40 (75.47%) patients died, 10 (18.87%) patients survived, and 3 patients (5.66%) were lost to follow-up. [26] Among the 40 dead patients, 38 patients died from rapid disease progression or tumor recurrence and 2 died of severe post-operative complications. Most of the relapses/recurrences (94.11%) occurred within 8 months, with a median time of 76 days from diagnosis. The 5-year OS was 18.44%, with younger age at diagnosis and higher stage patients had a relatively poor prognosis. Statistically significant differences were noted among patients treated with standard chemotherapy, total resection, and radiotherapy.
In a smaller case analysis of 14 children with MRT of the kidney treated in Paediatrics of Beijing Tongren Hospital from January 2010-December 2019, Li et al. reported a 4-year OS rate of 41.8%. Factors associated with a poor prognosis were age (younger than 24 months), a high Ki67 proliferation index (≥ 70%), and the presence of distant metastases. The lungs were the most common site of distant metastasis. [27]
Survival outcomes remained poor for those patients with higher stage disease treated on the most recent COG study AREN0321 (regimen UH1), a more intensive chemotherapy regimen than previously used in NWTSG trials. The 4-year event-free survival (EFS) and OS for the entire cohort were 23.1% and 33.3%, respectively. [28]
The 4-year OS for patients with MRT in AREN0321 was as follows:
-
Stage I - 2 patients (100%)
-
Stage II - 5 patients (100%)
-
Stage III - 24 patients (25%)
-
Stage IV - 9 patients (11.1%)
-
Stage V - 0 patients
Such data suggest that intensive therapy may benefit rare low stage MRT patients (Stage 1 and 2), but novel therapy is necessary for Stage 3 and 4 patients.
Complications
Complications may be related to tumoral progression or to treatment, as follows:
-
Complications related to tumoral progression: MRTs in the abdomen can rapidly progress, as can those at metastatic sites, including the lungs, liver, and brain. MRTs can be associated with tumoral hemorrhage and organ failure.
-
Complications related to treatment include the following:
- Hematologic complications: The major acute complication of chemotherapy for MRTs is myelosuppression, which places patients at risk for serious infections. Patients require frequent RBC and platelet transfusions.
- Renal complications: Patients may have renal tubular dysfunction, with wasting of protein, phosphorous, bicarbonate, and other electrolytes if platinum drugs or ifosfamide are used. The long-term prevalence of renal failure is unknown because MRTs are rare and the survival rate is low. Renal failure is uncommon in patients with unilateral Wilms tumor; however, patients with MRTs are treated intensively and with additional nephrotoxic drugs.
- Cardiac complications: Some treatment regimens for MRTs include anthracyclines, which can cause arrhythmias and congestive heart failure. Cardiac function should be monitored periodically.
- Gonadal complications: Ifosfamide and cyclophosphamide are associated with a risk of infertility.
- Secondary cancers: The risk of secondary cancers from chemotherapy and/or radiation, particularly in patients with a genetic rhabdoid cancer predisposition, remain unknown.
Patient Education
Patients and families should be educated about MRT and its aggressive biologic behavior.
Although families must be given hope for a cure, they must also be made aware of the unfavorable prognosis associated with MRTs, especially those presenting at an advanced stage. Families must also understand the risks of intensive chemotherapy and the signs and symptoms that require immediate medical attention.
Genetic counseling
Genetic counseling is highly recommended for all MRT affected families.
The incidence of germline deletions or missense mutations of SMARCB1 in infants and children with MRT approximates 15-30%. Families with more than one affected child have been reported; in 2 families, evidence of germline mosaicism was suggested because neither parent had a mutation in their own peripheral blood. The incidence and age distribution of cancer in individuals with inherited SMARCB1 mutations has not been formally studied, but adults without cancer have been shown to transmit the abnormal allele in at least 3 families, and individuals with germline perturbations of SMARCB1 are predisposed to malignant rhabdoid tumors of the kidney, soft tissues, and brain and may, in fact, present with more than one primary tumor.
Accumulating evidence suggests that individuals with a confirmed MRT should be evaluated for SMARCB1 expression in the tumor. Direct evaluation of the tumor by karyotyping, fluorescence in situ hybridization (FISH), or genomic microarray, with high-density single nucleotide polymorphism-based oligonucleotide arrays and multiplex ligation-dependent probe amplification as necessary, should be pursued to detect the mechanisms for biallelic silencing of SMARCB1 expression. Direct sequencing of SMARCB1 for missense mutations is recommended if abnormalities are not seen in both alleles. Evaluation of peripheral blood should follow tumor analysis. The finding of a chromosomal abnormality involving 22q or SMARCB1 missense mutation in the germline of an affected individual would then be followed by testing both parents. Because sibling recurrence is known to occur, testing of siblings, particularly those younger than 5 years, should be considered, even if bothparents are healthy.
Surveillance of individuals found to carry a constitutional SMARCB1 mutation for the development of CNS or abdominal MRT may be advisable (see Deterrence/Prevention).
-
Nonenhanced CT scan demonstrates linear and curvilinear calcifications outlining tumor lobules in a malignant rhabdoid tumor (MRT) (arrows). A hypoattenuating fluid collection surrounds and separates the lobules. These imaging features are seen with MRT more often than with other childhood renal neoplasms.
-
Contrast-enhanced CT scan demonstrates a subcapsular fluid collection (arrow) and the lobulated nature of a malignant rhabdoid tumor (MRT). Subcapsular fluid collections are more common with MRTs than with the other renal neoplasms that occur in children.
-
Histology of malignant rhabdoid tumors (MRTs). This photomicrograph shows the typical large malignant cells with large, vesicular nuclei, prominent red nucleoli, and abundant eosinophilic cytoplasm. Many tumor cells have a distinct, pale, rhabdoid inclusion in the cytoplasm. (Hematoxylin and eosin stain, original magnification x400).
-
INI1 immunohistochemistry stain shows diffuse loss of INI1 expression in tumor nuclei, with appropriate staining of intratumoral endothelial cells serving as the internal control (original magnification x400).
Tables
- Table 1. Acquired SMARCB1 Alterations in 200 Sporadic Rhabdoid Tumors (Adapted from Geller JI, Roth JJ, Biegel JA. Biology and Treatment of Rhabdoid Tumor. Crit Rev Oncog. 2015. 20 [3-4]:199-216)
- Table 1. One Ifosfamide-Carboplatin-Etoposide regimen for MRT
- Table 2. One Vincristine-Doxorubicin-Cyclophosphamide Regimen for MRT
Allele 1 Alteration |
Allele 2 Alteration |
Total |
|||
Mutation |
Partial Gene Deletion/Duplication |
Whole Gene Deletion |
CN-LOH |
||
Mutation |
8 (4%) |
1 (0.50%) |
58 (29%) |
27 (13.5%) |
94 (47%) |
Partial Gene Deletion/Duplication |
- |
5 (2.5%) |
14 (7%) |
11 (5.3%) |
30 (15%) |
Whole Gene Deletion |
- |
|
76 (38%) |
- |
76 (38%) |
Total |
8 (4%) |
6 (3%) |
148 (74%) |
38 (19%) |
200 (100%) |
Drug |
Dosage |
Route |
Schedule |
Carboplatin |
Target dose to the AUC of 6 mg/mL/min by using the Calvert equation |
IV |
Day 1 |
Etoposide |
3.3 mg/kg/dose or 100 mg/m2/dose |
IV |
Days 1, 2, and 3 |
Ifosfamide |
65 mg/kg/dose or 2 g/m2/dose |
IV |
Days 1, 2, and 3 |
Mesna |
16 mg/kg/dose or 500 mg/m2/dose |
IV |
Start immediately after and at 3 h, 6 h, and 9 h after ifosfamide |
Filgrastim G-CSF |
5 mcg/kg/dose |
SC |
Start 24 h after chemotherapy and continue until ANC recovers |
Drug |
Dosage |
Route |
Schedule |
Vincristine |
0.05 mg/kg/dose or 1.5 mg/m2/dose; not to exceed 2 mg/dose |
IV |
Days 1, 8, and 15 |
Doxorubicin |
1.2 mg/kg/dose or 37.5 mg/m2/dose |
IV |
Days 1 and 2 |
Cyclophosphamide |
60 mg/kg/dose or 1.8 g/m2/dose |
IV |
Day 1 |
Mesna |
15 mg/kg/dose or 450 mg/m2/dose |
IV |
Start immediately after and at 3, 6, and 9 h after cyclophosphamide |
Filgrastim G-CSF |
5 mcg/kg/dose |
SC |
Start 24 h after chemotherapy and continue until ANC recovers |






