Overview
Introduction
Meconium ileus is among the most common causes of intestinal obstruction in the newborn, accounting for 9-33% of neonatal intestinal obstructions. It is the earliest clinical manifestation of cystic fibrosis (CF) and occurs in either a simple or a complicated form in approximately 16-20% of patients who have CF, though it also occurs in patients who do not have CF.
In the simple form, thickened meconium begins to form in utero. It obstructs the midileum, causing proximal dilatation, bowel wall thickening, and congestion. Complicated meconium ileus may cause volvulus, atresia, necrosis, perforation, meconium peritonitis, and pseudocyst formation.
Clinically, CF is characterized by the following triad:
-
Chronic obstruction and infection of the respiratory tract
-
Exocrine pancreatic insufficiency
-
Elevated sweat chloride levels
A possible meconium ileus diagnosis should raise the suspicion of CF in the fetus. Antenatal diagnosis of meconium ileus can be confirmed in 2 groups. In the low-risk group, the diagnosis is suspected when routine prenatal ultrasonography reveals the sonographic appearances of meconium ileus. The high-risk group consists of all pregnancies subsequent to the birth of a child with CF. Parents of a child with CF are obligate carriers of a CF mutation.
Go to Cystic Fibrosis and Sinonasal Manifestations of Cystic Fibrosis for complete information on these topics.
Pathophysiology
Fetuses with CF have abnormal development of the pancreas and intestinal tract. Expression of the cystic fibrosis transmembrane conductance regulator gene (CFTR) can be detected in the pancreatic ductules at 18 weeks’ gestation. In patients with CF, abnormal pancreatic secretions obstruct the duct system, leading to autodigestion of the acinar cells, fatty replacement, and ultimately, fibrosis. Beginning in utero, this progressive process occurs variably over time.
Approximately two thirds of infants later diagnosed with CF by neonatal screening have pancreatic insufficiency at birth. Approximately 10% of patients with CF remain pancreatic sufficient and tend to have a milder course.
Indications
Surgical exploration is indicated for patients with progressive distention, signs of peritonitis, or clinical deterioration. Surgery is always indicated for complicated meconium ileus. Complicated meconium ileus requires resection more often than simple meconium ileus and always requires temporary stomas.
The following complications necessitate surgical management:
-
Persistent or worsening abdominal distension
-
Persistent bowel obstruction
-
Enlarging abdominal mass
-
Perforation
-
Meconium cyst formation with peritonitis
-
Bowel necrosis
Conditions associated with cystic fibrosis and meconium ileus
Complicated meconium ileus may cause volvulus, atresia, necrosis, perforation, meconium peritonitis, and pseudocyst formation. Infants with meconium ileus are at risk for cholestasis, particularly if they have received or are receiving total parenteral nutrition (TPN). Monitor alkaline phosphatase, alanine aminotransferase (ALT), aspartate aminotransferase (AST), and bilirubin levels weekly. An observational study of infants with CF and meconium ileus identified the following risk factors for poor outcomes: high blood immunoreactive trypsinogen levels, prenatally diagnosed intestinal obstruction, a severe post-surgical clinical picture, and early liver disease. [1]
Gastroesophageal reflux
Gastroesophageal reflux (GER) is more prevalent in patients with CF, and it may exacerbate the respiratory status of the patient with CF. Pathologic reflux (ie, endoscopic and histologic esophagitis) is present in more than 50% of patients with CF. Most CF patients have an abnormal quantity of reflux as defined by pH probe, and it has been reported with prominent respiratory symptoms. Early diagnosis and treatment of this condition are of prime importance if the complications of pathologic reflux are to be curtailed and respiratory function maximized.
The particular mechanism of GER in CF is unclear, but a number of factors may contribute to the increased susceptibility of this patient group to the development of pathologic GER.
First, most reflux episodes in CF occur during transient lower esophageal sphincter (LES) relaxations. These transient relaxations are increased during distension of the gastric fundus, a feature that can predispose these patients to reflux especially when receiving large supplemental bolus feeds. This cycle of events may be further exacerbated in the event of poor gastric emptying. Gastric emptying of liquids was initially thought to be delayed in patients with CF.
Second, the head-down posture adopted during chest physiotherapy places gastric liquid content in an optimum position at the LES for reflux in the event of a transient period of relaxation. Associated coughing and forced expiration, which both increase the abdominothoracic pressure gradient, also facilitates reflux action.
Last, medications such as theophylline and beta-adrenergic drugs, used in the treatment of respiratory disease in patients with CF, are known to decrease the resting tone in the LES and could conceivably facilitate reflux activity.
Biliary tract disease
Biliary tract disease is another important consideration. Gallbladder disease is prevalent in the CF population, with abnormal oral cholecystograms in 46% and cholelithiasis in 12%. Abnormalities described in patients with CF include a microgallbladder containing thick colorless “white bile” with occlusion of the cystic duct, gallstones, biliary dyskinesia, and sclerosing cholangitis.
Bile acid metabolism is disturbed in patients with pancreatic insufficiency who are not receiving adequate pancreatic enzyme supplementation. Bile acids are likely bound to malabsorbed fat and, as a result, are lost in feces, which in turn depletes the bile acid pool and supersaturates cholesterol in the gallbladder. This condition promotes stone formation.
Many patients with CF and gallbladder sludge or stones are asymptomatic, but approximately 4% have the classic symptomatology of cholecystitis. A laparoscopic cholecystectomy is the treatment of choice in such cases because postoperative pain and therefore pulmonary compromise are less with this technique than with the classic open technique. The role of cholecystectomy in patients with asymptomatic gallstones remains unclear.
Distal intestinal obstruction
Distal intestinal obstruction syndrome (DIOS), also referred to as meconium ileus equivalent, is a recurrent postneonatal partial or complete intestinal obstruction unique to patients with CF. Most cases occur in adolescents and adults, but all age groups can be affected, with an overall incidence of approximately 15%. [2]
The exact etiology of DIOS is unknown, but these patients are more likely to have a history of steatorrhea from pancreatic exocrine insufficiency despite adequate enzyme therapy. One study also showed that, of 27 patients with DIOS, 17 (63%) had a history of meconium ileus as an infant.
A number of aspects peculiar to the gastrointestinal (GI) function of CF patients may help, in part, to explain this syndrome. These include abnormal intestinal mucins, abnormal intraluminal water and electrolyte content, and inherently slow intestinal motility. The latter may be because neurotensin, a GI hormone that delays motility, is secreted from the distal ileum when unabsorbed fat reaches that location. Additional precipitating factors may include relative dehydration, especially in a postoperative period, inadequate enzyme supplementation, and changes in diet.
The cardinal features of DIOS are cramping abdominal pain, often localized to the right lower quadrant (RLQ), a palpable mass in the RLQ, and decreased frequency of defecation. Different degrees of obstruction are present, from partial, which is most common, to complete with vomiting, distension, and absolute constipation. Colicky pain may be provoked by meals, which may then result in anorexia as a method to avoid further pain.
Physical examination in uncomplicated DIOS usually reveals a tender mass in the RLQ with no evidence of peritonitis. No fecal impaction or dehydrated stool is noted on rectal examination, and the stool is heme negative.
The nonspecific nature of DIOS, with no pathognomonic radiologic features, means that an accurate diagnosis of abdominal pain in the patient with CF is not easy. Plain supine and erect abdominal radiography is still, however, the most helpful initial investigation when the diagnosis is suspected. This shows bubbly granular material in the right iliac fossa and variable degrees of small-bowel obstruction, ie, air-fluid levels with proximal small-bowel dilatation. Plain radiography supports, but does not prove, the diagnosis.
Inspissated material in the right iliac fossa can also be demonstrated with a water-soluble contrast enema. In doing so, intussusception can be excluded, and the investigation itself may prove therapeutic in some cases of DIOS.
Particular difficulty is faced in the event of partial small-bowel obstruction caused by adhesions from previous abdominal surgery or appendiceal disease, which occurs in 1.5-2% of patients with CF (a lower incidence than the 8.6% noted in the general population).
Abdominal pain is a common symptom in patients with CF, and, because these patients are often already being treated with antibiotics and steroids, the classic clinical signs and symptoms of appendicitis may be masked and the critical diagnosis missed. This results in a high incidence of perforation and substantial morbidity in this patient group. Despite the blunting of clinical signs, evidence of pyrexia and a leukocytosis may still be present.
Depending on the appendix location, a contrast enema may show deformity of the cecum with an associated mass effect and not the typical inspissated material features of DIOS. Abdominal ultrasonography or, if necessary, computed tomography (CT) scanning shows free fluid or an abscess collection in the region of the cecum. In such cases, treatment should then proceed with appendectomy. If the diagnosis is still in doubt, the surgeon could opt to start with a laparoscopic investigation and then proceed appropriately in light of the findings.
In the absence of partial small-bowel obstruction due to adhesions, appendiceal disease, or complete obstruction, DIOS is suitable for a trial of medical management. After adequate rehydration, a balanced polyethylene glycol–electrolyte solution may be administered orally or via nasogastric tube. The dosage is 20-40 mL/kg/h with a maximum of 1200 mL/h. Prokinetic agents, such as metoclopramide, can be used to limit the amount of nausea and bloating.
Successful treatment is judged by the passage of stool, resolution of symptoms, and the disappearance of a previously palpable right iliac fossa mass. Sequential plain abdominal radiography helps to document the resolution of DIOS, but, if symptoms persist, the differential diagnoses already outlined must be reconsidered.
The use of enemas at the diagnostic stage is outlined. Contrast enemas should also be used for patients with emesis due to DIOS after placement of a nasogastric tube for gastric suction. As long as the patient remains clinically stable, the contrast enemas may be repeated at intervals of several hours over several days. However, careful monitoring of the patient must be initiated before, during, and after the procedure because large fluid and electrolyte shifts can be induced by the contrast material.
When complete obstruction or evidence of peritonitis is present, surgical intervention is necessary and all oral or rectal therapies are contraindicated. A nasogastric tube should be passed to help with decompression, and adequate resuscitative measures should be initiated. At laparotomy, the bowel wall feels thickened and filled with tenacious material. It can be decompressed and irrigated with diatrizoate meglumine, usually via a small catheter placed through the appendix stump. An irrigating tube may also be left in situ for postoperative bowel irrigation.
Gastrointestinal neoplasms
The overall risk of cancer in patients with CF is similar to that of the general population; however, risk of gastrointestinal neoplasms is increased. These include tumors of the esophagus, stomach, small intestine, large intestine, liver or biliary tract, and pancreas.
The differential localization and expression of CFTR may play a role in the neoplastic disease process. Furthermore, increased cellular turnover in response to the persistent irritation of GER, gallstones, or steatorrhea in digestive tract organs may also offer an explanation to these findings.
Fibrosing colonopathy
Fibrosing colonopathy is a newly described entity in children with CF. A longitudinal study showed that, of the children with CF who developed fibrosing colonopathy, 89% (8) of patients underwent surgery, and 63% (5) of them received a subtotal colectomy. Findings at laparotomy in children with CF who presented with presumed DIOS that did not respond to medical therapy included colonic strictures with histopathological changes of postischemic ulceration repair, with mucosal and submucosal fibrosis, destruction of the muscularis mucosa, and eosinophilia.
In some patients, a change from conventional enteric-coated pancreatic enzymes to high-strength products 12-15 months before presentation has been described.
The diagnosis of fibrosing colonopathy should be considered in patients with CF who have been exposed to high doses of pancreatic enzymes and present with symptoms of abdominal pain, distension, chylous ascites, change in bowel habit, or failure to thrive. Continued diarrhea may also be a prominent feature, which unfortunately may prompt the family to increase supplemental enzymes further. On occasion, the diarrhea may be bloody.
A barium enema may reveal mucosal irregularity, loss of haustral markings with a foreshortened colon with varying degrees of stricture formation. In some cases, the whole colon has been involved. Colonoscopy may show an erythematous mucosa and areas of narrowing, from which taking multiple forceps-pinch biopsies is advisable.
Initial management should reduce enzyme dosage to the recommended levels of 500-2500 lipase units/kg per meal. This should be accompanied with adequate nutritional supplementation, which may be enteral elemental feeding or even TPN for a time. Those patients who show signs of unrelenting failure to thrive, obstruction, uncontrollable diarrhea, or chylous ascites then need surgical intervention.
When elective surgery is planned for patients with intractable symptoms, gentle bowel preparation can be administered preoperatively. The aim of surgical intervention is to resect the affected bowel and make a primary anastomosis. Unfortunately, this is not possible in the event of pancolonic or rectal involvement, and, as a result, the patient requires an ostomy. This is often the safest option; patients and parents must be fully aware and prepared for it preoperatively.
Whether this condition completely resolves with a reduction in enzyme dosage and surgical resection is unclear, so the operated group also requires regular follow-up for any signs of deterioration.
Rectal prolapse
Rectal prolapse occurs in approximately 20-23% of patients with CF. The initial prolapse occurs most commonly in patients aged 1-3 years and can be recurrent in nature. It may also be the sole presenting feature of a new patient with CF in about 4-8% of all cases.
Factors that directly predispose this group to prolapse include constipation, diarrhea with increased frequency and volume of movement, malabsorption, and colonic distention. Indirect contributors relate to increased intra-abdominal pressure caused by coughing or pulmonary hyperinflation.
Initial management involves manually reducing the prolapse. Medical management to maximize fat absorption then aids the overall control. Reassure parents that the number of prolapse episodes is likely to reduce with age. However, further intervention is warranted in a small group when they have persistent pain or incontinence with each episode of prolapse.
The acute prolapse is easily reduced if action is taken promptly before edema formation. Parents can be taught to grasp the herniated bowel with the fingertips of a gloved hand and apply circumferential pressure with an inward push. Sustained pressure may be required to achieve full reduction. If prolapse immediately recurs, then the buttocks can be strapped together with adhesive tape for 7-14 days.
Recurrent prolapse can be treated by means of a rectal submucosal injection, performed with general anesthesia after the rectum has been emptied with a suppository. With the patient in the lithotomy position, the needle is inserted through the skin just outside the mucocutaneous junction and guided into position by a finger placed in the rectum. As the needle is slowly withdrawn, 2-3 mL of 5% phenol in almond oil or hypertonic sodium chloride solution (30%) is injected in a linear track into 4 different quadrants.
A single treatment controls approximately 90% of cases. Linear electrocauterization in the 4 quadrants has also been described to produce a perirectal inflammation. This technique requires a longer hospital stay and may be complicated by rectal bleeding and/or rectal stenosis.
When all conservative options are exhausted, a surgical approach may then be considered. However, many different operations have been described to control rectal prolapse. Through a transabdominal approach, the rectum can be fixed to the hollow of the sacrum by a prosthetic or fascia lata graft sutured to the bowel and the presacral fascia, thus creating a new pelvic floor.
Other operations include rectal suspension and levator ani muscle repair through a posterior sagittal approach. The diversity of options highlights the unsatisfactory results often achieved in these difficult cases.
Preparation
Initial medical management
Manage both simple and complicated meconium ileus in newborns as an intestinal obstruction. Perform resuscitative measures, including mechanical respiratory support, if necessary. Initiate intravenous (IV) hydration with gastric decompression, evaluate and correct any coagulation disorders, and begin empiric antibiotic coverage. Immediately obtain a surgical evaluation when meconium ileus is suspected or diagnosed.
Diatrizoate meglumine enema
In 1969, Noblett introduced the use of Gastrografin enemas to treat 4 infants with meconium ileus. [3] Variations on this approach are now the preferred initial method to treat uncomplicated meconium ileus.
Gastrografin, marketed by Bristol-Myers Squibb of Princeton, NJ, is meglumine diatrizoate, a hyperosmolar, water-soluble, radiopaque solution containing 0.1% polysorbate 80 (Tween 80) and 37% organically bound iodine. The solution’s osmolarity is 1900 mOsm/L.
The reported success rate of Gastrografin enemas for patients with uncomplicated meconium ileus is 63-83%. In a 2005 analysis, the success rate was only 35% (15 out of 42 patients) for simple meconium ileus relief, whereas 64.2% (27) of patients needed surgery to relieve the obstruction. [4] See the images below.
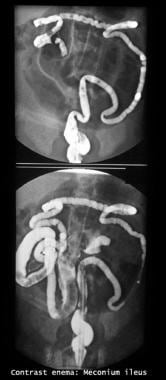 Gastrografin enema study shows filling defects in the terminal ileum and cecum. Also note the microcolon (transverse and descending colon).
Gastrografin enema study shows filling defects in the terminal ileum and cecum. Also note the microcolon (transverse and descending colon).
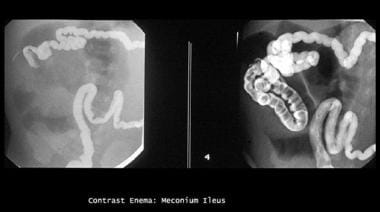 Enema study shows microcolon and contrast material outlining a terminal ileum packed with (meconium) filling defects.
Enema study shows microcolon and contrast material outlining a terminal ileum packed with (meconium) filling defects.
Noblett’s criteria for proceeding with this therapy require the following:
-
The initial diagnostic contrast enema must exclude other causes of neonatal distal intestinal obstruction.
-
The infant must show signs of uncomplicated meconium ileus and no clinical or radiologic evidence of complicating factors (eg, volvulus, gangrene, perforation, peritonitis, atresia of the small bowel).
-
The infant should be well prepared for the enema, with adequate fluid and electrolyte replacement and correction of hypothermia.
-
The enema must be performed under fluoroscopic control.
-
IV antibiotics should be administered.
-
Close surgical supervision is imperative from the initial evaluation through the hospital course.
Upon instillation, fluid is drawn into the intestinal lumen to hydrate and soften the meconium mass. Both transient osmotic diarrhea and diuresis follow. Adequate resuscitation and hydration in anticipation of these fluid losses is paramount.
Under fluoroscopic control, infuse a 25-50% solution of Gastrografin slowly at low hydrostatic pressure through a catheter inserted into the rectum. Avoid balloon inflation to minimize the risk of rectal perforation. To help deconcentrate the inspissated meconium, 1% N- acetylcysteine may be added to the enema solution. This procedure usually prompts rapid passage of semiliquid meconium, which continues for 24-48 hours.
Upon completion, withdraw the catheter and obtain an abdominal radiograph to exclude perforation. Return the infant to the neonatal care unit for intensive monitoring and fluid resuscitation. Obtain radiographs in 8-12 hours, or as clinically indicated, to confirm evacuation of the obstruction and to exclude late perforation.
A second enema may be necessary for nonoperative management of meconium ileus if evacuation is incomplete or if the first attempt at Gastrografin evacuation does not reflux contrast into dilated bowel. If necessary, serial Gastrografin enemas can be performed at 6- to 24-hour intervals.
Surgical exploration is indicated for patients with progressive distention, signs of peritonitis, or clinical deterioration.
Following successful evacuation and resuscitation, Noblett suggests administering a 10% N- acetylcysteine solution (5 mL q6h) through a nasogastric tube to liquefy upper GI secretions.
Feedings, including supplemental pancreatic enzymes for infants with confirmed CF, may be initiated when signs of obstruction have subsided, usually within 48 hours.
Potential complications include perforation, hypovolemic shock, and ischemia.
Rectal perforation can be avoided by carefully placing the catheter under fluoroscopic guidance and avoiding inflating balloon-tipped catheters. In 1987, Ein et al reported a 23% perforation rate in patients treated with inflated balloon catheters. [5] Early perforation that occurs during enema administration is usually apparent under fluoroscopy. Perforation risk increases with repeated enemas.
Late perforation can occur 12-48 hours after the enema. Potential causes include severe bowel distention by fluid osmotically drawn into the intestine (the apparent etiology in experimental models) or by direct injury to the bowel mucosa by the contrast medium. Delayed perforation associated with extensive bowel necrosis has been reported. The pathogenesis of intestinal perforation associated with necrotizing enterocolitis is believed to be the ischemia produced by intestinal distention.
Hypovolemic shock is a profound risk when hypertonic enemas are delivered. Ischemia caused by overdistention is worsened by hypoperfusion; this hypoperfusion is caused by the hypovolemia that results from poor fluid resuscitation. Adequate fluid resuscitation (ie, 150 mL/kg/d minimum), including anticipated fluid losses from osmotic diarrhea and diuresis, is mandatory.
Technique
Overview
A number of surgical approaches to treat uncomplicated meconium ileus have been proposed over the years; variable success rates have been achieved. Individualize the approach for each infant.
The goal of operative management in simple uncomplicated meconium ileus is to evacuate meconium from the intestine while preserving maximal intestinal length.
Surgery is always indicated for complicated meconium ileus. Complicated meconium ileus necessitates resection more often than simple meconium ileus does (see Indications) and may necessitate the use of temporary stomas.
Resection and enterostomy
Several variations of the technique used by Hiatt and Wilson [6] have involved placing indwelling ostomy tubes for postoperative bowel irrigation decompression and/or feeding. In 1970, O’Neill et al described success with tube enterostomy, with and without resection. [7] In 1981, Harberg et al described a similar procedure using a T-tube enterostomy. [8]
In either situation, begin irrigations on the first postoperative day; after successfully clearing the obstruction (ie, 7-14 d), the physician may remove the tube and allow the enterocutaneous fistula to close spontaneously.
Subsequent surgical techniques have revolved around resection, anastomosis, and enterostomy, through which postoperative irrigations can be delivered. The Mikulicz double-barreled enterostomy, first reported by Gross in 1953, has the following distinct advantages:
-
The procedures reduce operating and anesthetic times because complete evacuation of inspissated meconium is unnecessary.
-
The procedures avoid intra-abdominal anastomosis, which eliminates the risk of anastomotic leakage.
-
The bowel can be opened after complete closure of the abdominal wound; this reduces the risk of intraperitoneal contamination.
After the operation, solubilizing agents can be administered through the proximal or distal limbs of the stoma, per rectum, or via a nasogastric tube. In the classic description, a crushing clamp may be applied to the 2 limbs to create continuity for distal flow of intestinal fluids.
The disadvantages of this and other procedures involving resection and stoma(s) are potential postoperative fluid losses through high-volume stomas, bowel shortening by resection, and the need for a second procedure to reestablish intestinal continuity.
A distal chimney enterostomy, as described by Bishop and Koop in 1957, [9] involves resection with anastomosis between the end of the proximal segment and the side of the distal segment of bowel, approximately 4 cm from the opening of the distal segment. The open end is brought out as the ileostomy. This technique allows normal gastrointestinal (GI) transit while providing a means for managing distal obstruction through the ileostomy, should it occur.
The reverse of the Bishop-Koop enterostomy is the proximal enterostomy, described by Santulli and Blanc in 1961. [10] In this technique, after resection, the end of the distal limb is anastomosed to the side of the proximal limb. The end of the proximal limb is brought out as the enterostomy. This arrangement enhances proximal irrigation and decompression, and evacuation of the proximal small bowel at the time of surgery is unnecessary.
As with distal chimney enterostomy, a catheter is placed for access to the distal limb. The catheter exits through the stoma to provide a means to irrigate the distal bowel. This technique’s apparent disadvantage is the presence of a high-output stoma and the inherent risk of dehydration.
Take care to replenish fluids, electrolytes, and nutrients in accordance with the stomal output. Reinstallation of stomal output from the proximal to the distal limb often is performed via the indwelling catheter.
Resection and primary anastomosis
Resection with primary anastomosis was suggested first by Swenson in 1962. [11] Initially, this technique had difficulties and complications with leakage from the anastomosis. More recently, Chappell in 1977 [12] and Mabogunje et al in 1982 [13] have reported improved results. These authors emphasize the necessity of adequately resecting the compromised bowel, completely evacuating proximal and distal meconium, and preserving an adequate blood supply to the anastomosis.
Modified gross technique
The author prefers a modification of the technique originally described by Gross in 1953 [14] to manage infants with uncomplicated meconium ileus.
This modified technique begins by performing a celiotomy with a muscle-sparing horizontal incision just above the umbilicus. Upon exploration, a decision is made, based on the viability and length of the bowel, either to create an enterotomy for irrigation and evacuation of the meconium or to resect the segment of impacted intestine.
The author then creates side-by-side separate enterostomies without creating a common wall. Stomas are placed within the abdominal incision to the right; these may be covered with a single ostomy collecting device. Postoperatively, each stoma may be irrigated to remove residual meconium. [15]
Instillation of dilute enteral feedings high in glutamine, via the distal stoma, may also be performed to stimulate growth of the unused distal bowel. Intestinal continuity is usually restored within 6 weeks if bowel function resumes and the infant tolerates oral feedings.
Transplantation for associated pathology
Owing to associated hepatic pathology, a liver transplant may be indicated. A study of 10 patients with CF who underwent orthotopic liver transplantation (OLTX) found a mortality rate of 40%; all 4 patients who died had a history of meconium ileus. Of the 6 patients who survived; 4 (67%) had a history of meconium ileus. [16]
OLTX mortality was also associated with worse nutrition and development, a need for preoperative pancreatic enzymes, a higher incidence of pancreatic insufficiency, transplantation at an older age, and, probably most important, a longer waiting time until transplantation.
A case study of a 7-month-old boy with CF who needed a new liver and small intestine showed that early multivisceral transplantation can be performed safely. The boy weighed 6 kg (< 5th percentile at age of transplantation) and gained weight to 12 kg (75th percentile at age 3.6 y). [17]
Overall, the intestinal transplant survival rate is 77% for the first year and 64% at 5 years, whereas the 1-year graft survival rate is 67%, and the 5-year graft survival rate is 37%. Additionally, 87% of patients who receive intestinal transplant no longer require total parenteral nutrition (TPN).
Post-Procedure
Postoperative care
Initial postoperative management involves ongoing resuscitation. Carefully replace the fluid losses caused by surgery and by preoperative diuresis and diarrhea (if a Gastrografin enema was attempted). Adjust ongoing maintenance fluids and replace insensible fluids lost, as well as gastrointestinal (GI) losses (ie, losses from nasogastric suction and ileostomy).
Instillation of N -acetylcysteine via a nasogastric tube or an ileostomy helps solubilize residual meconium.
As soon as possible (ie, 6 wk) close the stomas placed in the course of surgical management to help avoid prolonged problems with fluid, electrolyte, and nutritional losses.
Nutritional management
Infants with uncomplicated meconium ileus and cystic fibrosis (CF) may receive breast milk or routine infant formula, enzymes, and vitamins. Use caution when prescribing enteric enzyme medication to patients with meconium ileus and CF. Reported complications include fibrosing colonopathy from excessive enzyme doses and distal intestinal obstruction syndrome (DIOS). Generic substitutions for proprietary medications have also been associated with treatment failures.
Patients who have a complicated surgical course require either continuous enteral feedings or total parenteral nutrition (TPN). The author recommends predigested infant formulas (eg, Alimentum, Pregestimil), for enteral feeding.
Prestenotic dilation of the small bowel caused by a meconium obstruction theoretically could cause mucosal damage that, in turn, could contribute to poor peristalsis or malabsorption. Patients who have had complicated meconium ileus or undergone a sizable bowel resection and who are fed enterally may tolerate continuous feedings better than bolus feedings.
Because bowel mucosa may or may not be damaged by stasis, begin feedings with predigested diluted formula, usually half strength, at low volume. Once this diluted formula is well tolerated, the physician may increase formula strength and then volume. During this process, look for signs of feeding intolerance (eg, abdominal distention, heme-positive stools, increasing emesis).
Once oral feedings begin, administer oral pancreatic enzymes, even with predigested formulas, starting at 2000-4000 lipase units per 120 mL of full-strength formula. For example, an infant who weighs 2.5 kg and who is receiving formula at 4 mL/kg/h should receive half of a 4000-lipase Pancrease capsule orally every 12 hours. Capsules containing enteric-coated microspheres can be opened. Capsule contents can be mixed with applesauce and administered orally.
Do not crush the microcapsules, because crushing exposes the enzymes to stomach acid during oral administration, which destroys the enzymes. Uncrushed pancreatic enzymes should be administered even with formulas that contain medium-chain triglyceride (MCT) oil.
If pancreatic enzymes cause skin breakdown, a zinc oxide ointment (Desitin) can be applied to perianal skin.
Expected outcomes
Hiatt and Wilson reported the first survivors with meconium ileus in 1948, [6] and early series reported 50-67% mortality rates. The advent of improved nonoperative and operative treatments, nutritional support, and treatment of bacterial infection have combined to improve reported survival rates for infants with both complicated and simple meconium ileus to 85-100%. Of the fatalities due to meconium ileus, Escobar et al showed 8 (9%) deaths over 32 years, with 5 of these patients having complicated meconium ileus and 3 having simple meconium ileus. [4]
However, patients with CF who have a history of meconium ileus have significantly worse pulmonary outcomes after age 8-10 years, as measured by forced expiratory volume in 1 sec (FEV1), forced vital capacity (FVC), forced expiratory flow at 25-75% (FEF25-75), and total lung capacity (TLC). Whether the meconium ileus was treated surgically or medically did not change future pulmonary status.
Overall, patients with CF and meconium ileus have worse lung function and a higher rate of obstructive disease than patients with CF who do not have a history of meconium ileus. These data support meconium ileus as being a different phenotype in CF.
An epidemiologic study of 27,703 patients, which used the age of CF diagnosis and the initial disease presentation to build survival models and to demonstrate disease severity in isolated patients with CF, revealed that in comparison with the SCREEN group diagnosed by a newborn screening program, patients diagnosed by meconium ileus had statistically significant higher risks of a shorter lifespan, acquisition of Pseudomonas aeruginosa infection, and a trend toward FEV1 below 70%. [18]
Additionally, each 1-year increase in age at diagnosis resulted in a 3% decrease in risk of shortened lifespan, a 5% decrease in acquiring P aeruginosa infection, and a 3% decreased risk of FEV1 less than 70%. [18]
Prenatal diagnosis of meconium ileus with obstetrical ultrasonography, coupled with improved biochemical and molecular techniques for diagnosis of CF, now enable a perinatal team to counsel a family about the likelihood of CF in the fetus. Prenatal diagnosis also allows physicians to monitor and manage an affected fetus to ensure an optimal outcome. Advances in neonatal care and surgical procedures for infants with CF complicated by meconium ileus have greatly improved survival rates. [19]
Complications
Infants who have had significant (ie, >33%) bowel resection may be difficult to manage, especially if the ileocecal valve has been resected. In addition, an ileostomy may lead to excessive fluid and sodium losses. Take down ostomies as soon as possible. In the interim, if access to the distal defunctionalized bowel is feasible, administer ostomy-drip feeds of glutamine-enriched formula at low volumes to enhance bowel growth and to help prevent bacterial translocation.
Gastric acid hypersecretion occurs in patients with short-bowel syndrome. The acidic intestinal environment inactivates pancreatic enzymes and prevents dissolution of enteric-coated microcapsules. Histamine 2 receptor blockers may be used as an adjunct to pancreatic enzyme therapy in patients who have had significant bowel resections.
Patients with the double burden of excessive sweat and intestinal sodium losses may require compensation for total body sodium deficit. Measure urine sodium levels in infants with ileostomies, especially those who do not grow, even if serum sodium levels are within the reference range. Infants with urine sodium levels less than 10 mEq/L need sodium (and possibly bicarbonate) supplementation.
Various pulmonary issues may arise. Although clinical lung disease usually does not develop early, mucus plugging and atelectasis can occur. Immediately postoperatively, initiate vigorous prophylactic pulmonary care with chest physiotherapy. Do not use the head-down position, because this increases the risk of gastroesophageal reflux (GER) and aspiration. Infants should receive nebulized albuterol (2.5 mg bid), followed by chest physiotherapy. Prophylactic antibiotics are unnecessary. Antibiotic therapy, if needed, should be based on respiratory tract cultures.
Future and controversies
Advances in perinatal diagnosis and management of meconium ileus and CF, combined with greater understanding of the cystic fibrosis transmembrane conductance regulator (CFTR) protein, [20, 21] have vastly improved the outlook for affected infants. Continued successful care for these patients depends on prenatal diagnosis, multidisciplinary care, and innovative antenatal therapy strategies.
The ability to detect both meconium ileus and CF prenatally means physicians should begin to consider strategies that would prevent simple meconium ileus from progressing to complex meconium ileus. The creation of mouse models for CF provides a unique opportunity to study the basic pathophysiology of meconium ileus. This knowledge should enable the development and prospective evaluation of new treatments for this disorder. Future prenatal interventions such as gene therapy may even prevent meconium ileus.
One example of such a gene therapy technique has been successfully applied in sheep. Using an ultrasound-guided injection into the stomach, David et al demonstrated the ability to transfer a gene to a fetal GI tract using a first-generation replication-deficient adenoviral vector. [22] If gene-transfer technology eventually proves safe and effective in humans, another door for prevention would open to eradicate or at least lessen the problems associated with inspissated meconium and other postnatal GI pathologies in CF.
Overall, future goals should include discoveries of new ways to reduce the perinatal complications that increase morbidity, mortality, and medical care costs.
-
Gastrografin enema study shows filling defects in the terminal ileum and cecum. Also note the microcolon (transverse and descending colon).
-
Enema study shows microcolon and contrast material outlining a terminal ileum packed with (meconium) filling defects.