Practice Essentials
Esophageal atresia refers to a congenitally interrupted esophagus. [1] One or more fistulae may be present between the anomalous esophagus and the trachea. The lack of esophageal patency prevents swallowing. In addition to preventing normal feeding, this problem may cause infants to aspirate and literally drown in their own saliva, which quickly overflows the upper pouch of the obstructed esophagus. If a tracheoesophageal fistula (TEF) is present, fluid (either saliva from above or gastric secretions from below) may flow directly into the tracheobronchial tree.
The condition was first described anecdotally in the 17th century. In 1670, Durston described the first case of esophageal atresia in one conjoined twin. In 1696, Gibson provided the first description of esophageal atresia with a distal TEF. In 1862, Hirschsprung (a famous pediatrician from Copenhagen) described 14 cases of esophageal atresia. In 1898, Hoffman attempted primary repair of the defect but was not successful and resorted to the placement of a gastrostomy.
At the start of the 20th century, surgeons were theorizing about how the lesion could be repaired. In 1939 and 1940, Ladd of Boston and Lever of Minnesota first achieved surgical success in stages; success meant that the affected children survived and skin-lined pharyngogastric conduits were eventually constructed. In 1941, Haight of Michigan successfully repaired esophageal atresia in a 12-day-old baby using a primary single-stage left-side extrapleural approach. Subsequent to that child's survival and with advances in surgical and anesthetic techniques, esophageal atresia is now regarded as an eminently correctable congenital lesion.
The treatment plan for each baby must be individualized. (See Treatment.) Prognostic classifications (eg, the Waterston, Spitz, and Poenaru prognostic classification systems) can provide guidance in patients with multiple problems and help determine the indications for and timing of surgical repair, but early and decisive identification of the most life-threatening anomaly is essential. Surgical approaches to treatment vary according to surgeons' preferences and variations in pathologic anatomy.
For patient education resources, see the Esophagus, Stomach, and Intestine Center and Procedures Center, as well as Choking and Bronchoscopy.
Pathophysiology
The variants of esophageal atresia have been described using many anatomic classification systems. To avoid ambiguity, the clinician should use a narrative description. Nevertheless, Gross of Boston described the classification system that is most often cited (see the image below). [2]
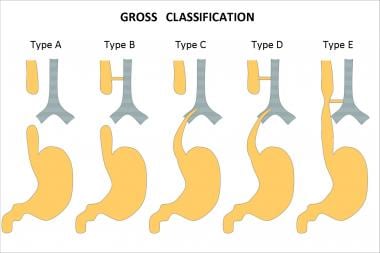
According to the system formulated by Gross, the types of esophageal atresia and their approximate incidence in all infants born with esophageal anomalies are as follows:
-
Type A - Esophageal atresia without fistula or so-called pure esophageal atresia (10%)
-
Type B - Esophageal atresia with proximal TEF (< 1%)
-
Type C - Esophageal atresia with distal TEF (85%)
-
Type D - Esophageal atresia with proximal and distal TEFs (< 1%)
-
Type E - TEF without esophageal atresia or so-called H-type fistula (4%)
-
Type F - Congenital esophageal stenosis (< 1%) (not discussed in this article)
A fetus with esophageal atresia cannot effectively swallow amniotic fluid, especially when TEF is absent. [3] In a fetus with esophageal atresia and a distal TEF, some amniotic fluid presumably flows through the trachea and down the fistula to the gut. Polyhydramnios may be the result of this change in the recycling of amniotic fluid through the fetus. Polyhydramnios, in turn, may lead to premature labor. The fetus also appears to derive some nutritional benefit from the ingestion of amniotic fluid; thus, fetuses with esophageal atresia may be small for their gestational age.
The neonate with esophageal atresia cannot swallow and drools copious amounts of saliva. Aspiration of saliva or milk, if the baby is allowed to suckle, can lead to an aspiration pneumonitis. In a baby with esophageal atresia and a distal TEF, the lungs may be exposed to gastric secretions. Also, air from the trachea can pass down the distal fistula when the baby cries, strains, or receives ventilation. [4] This condition can lead to an acute gastric perforation, which is often lethal.
Prerepair esophageal manometric studies have revealed that the distal esophagus in esophageal atresia is essentially dysmotile, with poor or absent propagating peristaltic waves. This condition results in variable degrees of dysphagia after the repair and contributes to gastroesophageal reflux (GER).
The trachea is also affected by the disordered embryogenesis in esophageal atresia. The membranous part of the trachea, the pars membranacea, is often wide and imparts a cross-sectional D shape to the trachea, as opposed to the usual C shape. These changes cause secondary anteroposterior structural weakening of the trachea, or tracheomalacia.
This weakening can result in a sonorous cough as the intrathoracic trachea resonates and partially collapses with forceful expiration. Secretions can be difficult to clear and may lead to frequent pneumonias. Also, the trachea can partially collapse during feeding, after repair, or with episodes of GER; this partial collapse can lead to ineffective respiration; hypoxia; and, somewhat inexplicably, apnea.
Etiology
No human teratogens that cause esophageal atresia are known. Esophageal atresia that occurs in families has been reported. A 2% risk of recurrence is present when a sibling is affected. The occasional association of esophageal atresia with trisomies 21, 13, and 18 further suggests genetic causation. Also, twinning occurs about six times more frequently in patients with esophageal atresia than in those without the condition.
Most authorities believe that the development of esophageal atresia has a nongenetic basis. [5] Debate about the embryopathologic process of this condition continues, and little about it is known. The old His theory that lateral infoldings divide the foregut into the esophagus and trachea is attractively simple, but findings from human embryology studies do not support this theory.
In 1984, O'Rahilly proposed that a fixed cephalad point of tracheoesophageal separation is present, with the tracheobronchial and esophageal elements elongating in a caudal direction from this point. [6] This theory does not easily account for esophageal atresia but explains TEF as a deficiency or breakdown of esophageal mucosa, which occurs as the linear growth of the organ exceeds the cellular division of the esophageal epithelium.
In a 1987 report, Kluth eschewed the concept that tracheoesophageal septation has a key role in the development of esophageal atresia. [7] Instead, he based the embryopathologic process on the faulty development of the early, but already differentiated, trachea and esophagus, in which a dorsal fold comes to lie too far ventrally; thus, the early tracheoesophagus remains undivided. He also suggested that esophageal vascular events, ischemic events, or both may be causes in cases of esophageal atresia without fistula.
In 2001, Orford et al postulated that the ectopic, ventrally displaced location of the notochord in an embryo at 21 days' gestation can lead to a disruption of the gene locus, sonic hedgehog-signaled apoptosis in the developing foregut, and variants of esophageal atresia. [8] This situation may be due to various early gestation teratogenic influences such as twinning, toxin exposure, or possible abortion.
In 2003, Spilde et al reported esophageal atresia-TEF formations in the embryos of rat models of doxorubicin-induced teratogenesis. [9] Specific absences of certain fibroblast growth factor (FGF) elements have been reported, specifically FGF1 and the IIIb splice variant of the FGF2R receptor. [10] These specific FGF-signaling absences are postulated to allow the nonbranching development of the fistulous tract from the foregut, which then establishes continuity with the developing stomach.
Epidemiology
The incidence of esophageal atresia is 1 case in 3000-4500 births. This frequency may be decreasing for unknown reasons. [11] Internationally, the highest incidence of this disorder is reported in Finland, where it is 1 case in 2500 births.
Prognosis
Statistics regarding mortality in esophageal atresia are constantly changing and improving. [12, 13, 14, 15] One must consider the classification system used in reporting such statistics.
Mortality relative to the Montreal classification is as follows [16] :
-
Class I - Mortality of 7.3%
-
Class II - Mortality of 69.2%
Mortality relative to the Spitz grouping is as follows [17] :
-
Group I - Mortality of 3%
-
Group II - Mortality of 41%
-
Group III - Mortality of 78%
Mortality relative to the Waterston categorization is as follows [18] :
-
Category A - Mortality of 0%
-
Category B - Mortality of 4%
-
Category C - Mortality of 11%
Fetuses with antenatal diagnoses of esophageal atresia seem to have a worse prognosis. [19] The cohort of babies in whom esophageal atresia is detected antenatally has a 75% mortality, whereas the cohort of babies in whom esophageal atresia is not detected antenatally has a 21% mortality. Babies who survive have varied morbidities related to any of the associated anomalies and complications. However, most children who undergo a successful repair of esophageal atresia are relatively healthy.
-
Esophageal atresia classification according to Gross.
-
This chest radiograph reveals esophageal atresia and distal tracheoesophageal fistula. Note Replogle tube in upper pouch and GI air below diaphragm.
-
This chest radiograph reveals esophageal atresia without tracheoesophageal fistula. Note absence of gas below diaphragm.
-
This radiograph reveals radius without radial ray deformity.
-
Contrast material has been administered, and probe has been placed through gastrostomy in this child with pure esophageal atresia. Air-filled upper pouch can be observed superiorly, with Replogle tube within it. This gapogram reveals very wide gap (>5 vertebral bodies), which requires esophageal replacement. This study is dynamic investigation, one in which surgeon and radiologist should be present to view real-time fluoroscopic images.
-
This postoperative contrast-enhanced radiograph reveals esophageal gastric tube replacement. Anastomosis to upper pouch is in chest. Linear staple line of tube can be observed.
-
This esophagogram was obtained using water-soluble contrast material 6 days after standard repair was performed. Chest tube is still in place. No leak is present. Waist observed here at site of recently performed anastomosis is usual and does not, at this stage, necessarily indicate stricture. Child went home and was eating well 3 days later.





