Practice Essentials
In 1679, Riverius recorded the first reported case of a congenital diaphragmatic hernia (CDH); this was after postmortem examination of a 24-year-old man. [1]
The first attempt at surgical repair of a CDH was by Nauman in 1888; the 19-year-old patient presented with acute respiratory distress and an acute abdomen, and a laparotomy was performed. In 1889, O'Dwyer carried out the first repair of CDH in an infant. The first successful repair was in 1905. The patient was aged 9 years, and Heidenhain reduced the hernia and closed the diaphragmatic defect through a midline laparotomy incision. Approximately 20 years later, Hedbolm reported a 58% mortality for patients undergoing surgical intervention for CDH.
In 1940, Ladd and Gross based their diagnosis of CDH on history, physical examination findings, and findings on chest radiography with or without a barium meal. [2] They advocated early surgical intervention (within the first 48 hours). Gross also described a two-stage closure of the abdominal wall in difficult cases; closure of skin and subcutaneous fascia at the initial surgery and closure of the abdominal wall 5-6 days later. In 1950, Koop and Johnson suggested the transthoracic approach as a means of closing the defect under more direct vision. [3]
As surgical expertise improved, innovative strategies were developed to address large diaphragmatic defects and agenesis of the hemidiaphragm. These techniques included the use of rotational muscle flaps, perirenal fascia, and synthetic patch repairs.
The exponential elucidation of the pathophysiology of CDH was instrumental in improving the survival rate in infants. CDH was no longer considered a primarily surgical disease but, rather, a disease associated with pulmonary hypoplasia, pulmonary hypertension, pulmonary immaturity, and an increased susceptibility of the lungs to ventilation-induced lung injury. This led to a delayed approach to surgical repair and to a gentle but more ingenious respiratory support.
Contemporary management of CDH emphasizes management of pulmonary hypoplasia and persistent pulmonary hypertension. Various gentle alveolar recruitment strategies are employed, and a nonurgent approach is taken to the operative treatment of CDH. Although the suggested window of opportunity for surgery is 24-48 hours after birth, surgical repair can often be safely delayed in stable patients, and the operation can be scheduled on a semielective basis. Urgent surgical repair is almost never necessary and may worsen the pulmonary hypertension.
For patient education resources, see the Digestive Disorders Center, as well as Hiatal Hernia.
Anatomy
The diaphragm is a musculotendinous structure that separates the thoracic cavity from the abdominal cavity. It is composed of a central nonmuscular portion (central tendon) surrounded by a muscular rim in addition to the right and left diaphragmatic crura. The right and left diaphragmatic crura are two muscular bands that originate from vertebral bodies L1-L3 and L1-L2, respectively. These muscular bands insert into the dorsomedial diaphragm.
Most diaphragmatic defects are posterolateral, with 85-90% of these occurring on the left. The label posterolateral may be a misnomer because it is frequently the case that much larger areas of the diaphragm are missing and only a posterior rim of muscle can be found. A hernial sac is present in 10-20% of cases.
The Morgagni defect occurs posterior to the sternum and results from failure of sternal and costal fibers to fuse at the site where the superior epigastric artery crosses the diaphragm. This defect is rare and is rarely a cause for surgery in the newborn.
Relevant embryology
The diaphragm is derived from four embryonic structures: the septum transversum, the pleuroperitoneal membranes, mesoderm of the body wall, and esophageal mesenchyme. After the folding of the fetal head at 4-5 weeks' gestation, the septum transversum comes to lie as a semicircular shelf, which separates the heart from the liver. The septum transversum does not completely separate the thoracic cavity from the peritoneal cavity but allows pericardioperitoneal canals to exist on either side of the esophagus.
During the fifth week of gestation, the pleuroperitoneal membranes develop along a line connecting the root of the 12th rib with the tips of the seventh to 12th ribs. The pleuroperitoneal membranes grow ventrally to fuse with the posterior margins of the septum transversum and the dorsal mesentery of the esophagus. Hence, at 6-7 weeks' gestation, the pleuroperitoneal canals are closed; the left closes after the right.
The mesentery of the esophagus condenses to form the left and right crura of the diaphragm, and the mesoderm of the body wall forms the outer rim of diaphragmatic muscle.
The posterolateral diaphragmatic defect is postulated to result from failure of closure of the pleuroperitoneal canals. The canal remains open when the intestines return to the abdomen at 10 weeks' gestation. Some intestine and other viscera enter the thorax and lead to compression of the developing lung at the crucial pseudoglandular stage and shifting of the mediastinum to the contralateral side. This causes compression of the heart and the contralateral lung as well.
In 1984, Iritani proposed a different concept of diaphragmatic development, suggesting that a posthepatic mesenchymal plate develops between the septum transversum and the pericardioperitoneal canals. [4] Lateral growth of this plate leads to closure of the pericardioperitoneal canals, and CDH results from a disturbance in growth of the posthepatic mesenchymal plate.
Pathophysiology
The pathophysiology of CDH involves pulmonary hypoplasia, pulmonary hypertension, [5] pulmonary immaturity, and potential deficiencies in the surfactant and antioxidant enzyme system.
Because of bowel herniation into the chest during crucial stages of lung development, airway divisions are limited to the 12th to 14th generation on the ipsilateral side and to the 16th to 18th generation on the contralateral side. Normal airway development results in 23-35 divisions. Because airspace development follows airway development, alveolarization is similarly reduced.
Development of the pulmonary arterial system parallels development of the bronchial tree, and therefore, fewer arterial branches are observed in CDH. Abnormal medial muscular hypertrophy is observed as far distally as the acinar arterioles, and the pulmonary vessels are more sensitive to stimuli of vasoconstriction. [6]
Pulmonary hypertension resulting from these arterial anomalies leads to right-to-left shunting at atrial and ductal levels. This persistent fetal circulation leads to right-side heart strain or failure and to the vicious circle of progressive hypoxemia, hypercarbia, acidosis, and pulmonary hypertension observed in the neonatal period.
The surfactant system is demonstrably deficient in the lamb model of CDH. [7] Postnatal administration of surfactant in these lambs is associated with dramatic increases in gas exchange, lung compliance, and pulmonary blood flow. However, in human neonates, reports on the status of the surfactant system are inconsistent. [8, 9]
Infants with CDH also have impairment of the pulmonary antioxidant enzyme system and are more susceptible to hyperoxia-induced injury.
In addition, a left ventricular smallness and hypoplasia are observed with CDH. This is believed to arise from decreased in-utero blood flow to the left ventricle, the mechanical compression of the herniated viscus similar to that observed in the lungs, and/or a primary yet unidentified developmental defect that simultaneously causes the diaphragmatic hernia and the lung problems.
Etiology
The genetic factors responsible for the development of CDH remain to be defined. [10, 11] Wide variations have been noted in the reported prevalence of chromosomal abnormalities (7-31%) in patients with CDH. The prevalence is higher in cases of CDH associated with other defects. [12] Familial occurrence has been noted in fewer than 2% of cases.
The role of drugs and environmental chemicals in the development of CDH is uncertain, but nitrofen, quinine, thalidomide, phenmetrazine, and polybrominated diphenyls have been used to induce CDH in various species. Investigations are exploring the link between CDH and defects in the retinoid signaling pathway in experimental models.
A small (N = 40) study from Japan found maternal dietary intake of vitamin A during pregnancy to be inversely associated with CDH in neonates. [13]
Epidemiology
CDH is generally considered to occur in approximately 1 per 3000 live births, though both lower and higher figures have been cited. [12, 14, 10, 15]
The Congenital Diaphragmatic Hernia Study Group recorded a 63% survival rate in 1995-1996 based on data from 62 centers in North America, Europe, and Australia. [16] Survival rates are 60-90% for patients who present within the first few hours of life (see the image below). [17]
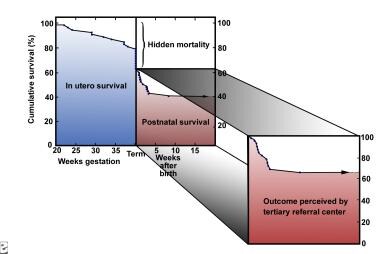 Graph illustrating the concept of the hidden mortality of congenital diaphragmatic hernia. Image courtesy of Michael Harrison, MD.
Graph illustrating the concept of the hidden mortality of congenital diaphragmatic hernia. Image courtesy of Michael Harrison, MD.
Prognosis
Long-term pulmonary disease depends on the degree of pulmonary hypoplasia, barotrauma, and volutrauma sustained in the neonatal period. Bronchopulmonary dysplasia and restrictive and/or obstructive lung disease may be observed.
Failure to thrive is often observed in the presence of optimal feeding regimes.
Functional and anatomic esophageal abnormalities are associated with significant gastroesophageal reflux (GER) in 40% of survivors. [6] Prophylactic fundoplication at the time of primary repair for infants requiring a patch repair is advocated by some team as a means of preventing growth disorders or failure to thrive in this subset of patients. Other patients who may go on to require fundoplication include the neurologically impaired and those with chronic lung disease. Most other infants outgrow GER. [18, 19]
The use of extracorporeal membrane oxygenation (ECMO), hyperventilation treatment, and ototoxic medication places this population at a higher risk for sensorineural hearing loss, as well as for neurodevelopmental abnormalities (ie, cognitive and developmental delay, cerebral palsy, seizure disorders, impaired vision).
Altered musculoskeletal development results in thoracic scoliosis, pectus deformities, and a decreased thoracic cavity on the affected side.
In late childhood and adolescence, learning disability, developmental disability, and attention deficit hyperactivity disorder (ADHD) are noted in approximately 50% of patients. In addition to cognitive and attention deficits, behavioral problems are seen. Only a third of these children require placement in a special educational class. [20]
-
Graph illustrating the concept of the hidden mortality of congenital diaphragmatic hernia. Image courtesy of Michael Harrison, MD.
-
Photograph of a one-day-old infant with congenital diaphragmatic hernia. Note the scaphoid abdomen. This occurs if significant visceral herniation into the chest is present.
-
Radiograph of an infant with congenital diaphragmatic hernia. Note shift of the mediastinum to the right, air-filled bowel in the left chest, and the position of the orogastric tube.
-
Newborn baby with congenital diaphragmatic hernia on venoarterial extracorporeal membrane oxygenation (ECMO). Note the arterial and venous cannulas connected to the bedside cardiovascular bypass machine.
-
Diagram illustrating the sheep model of PLUG, the trachea used for the fetal management of congenital diaphragmatic hernia. Image courtesy of Michael Harrison, MD.







