Background
Orthostatic intolerance is a confusing topic. Some of the confusion originates from recent appreciation of the condition's clinical variants, some originates from the emerging understanding of its diverse underlying pathophysiologies, and some originates from its nomenclature, which seems to change at least every year.
The term orthostasis literally means standing upright. Orthostatic intolerance (OI) may be defined as "the development of symptoms while upright, during standing that are relieved by recumbency." Although the use of a term such as orthostatic intolerance logically implies the presence of signs and symptoms when upright, variations in blood flow and blood pressure (BP) regulation are also found when supine or sitting, but these may require special equipment to detect and therefore may not be easily discernable until orthostatic stress becomes evident.
Standing successfully requires the interplay of blood volume, physical, neurologic, humoral, and vascular factors that compensate for the effects of gravitational venous pooling. Under ordinary conditions, acute humoral alterations have little to do with the initial response to standing upright but may play an important role during chronic orthostatic intolerance or relatively late during upright standing. Also, changes in such factors may affect resting or tonic responses and thus may influence overall vascular regulation through background effects.
If symptoms initiate while supine, then there is no OI. Transient OI is commonly experienced during dehydration or infectious disease. Typical signs and symptoms include: loss of consciousness or lesser cognitive deficits (memory loss, decreased reasoning and concentration); visual difficulties; lightheadedness; headache; fatigue; either increases of BP (hypertension), decreases of BP (hypotension); weakness; nausea and abdominal pain; sweating; tremulousness; and exercise intolerance. [1] Unless in harm's way (e.g., standing on a cliff), OI is not lethal. Some OI findings, such as nausea and sweating pertain directly to autonomic activation. However, loss of consciousness, severe lightheadedness, and neurocognitive loss relate to central nervous system (CNS) dysfunction and oblige recumbence.
CNS symptoms are produced by altered brain blood flow perhaps involving the brainstem. Cerebral Blood Flow (CBF) velocity is shown in the image below for two common forms of OI, vasovagal syncope (VVS) and postural tachycardia syndrome (POTS). Cerebral autoregulation may be compromised [2] as in POTS [3] and VVS [4] and may be reduced by hyperventilation and hypocapneic cerebral vasoconstriction. [5] Involuntary postural hyperventilation, mostly hyperpnea, is observed in all VVS patients [6] and 50% of POTS patients in our laboratory. [7] Trigeminal, sympathetic, or parasympathetic nerve activity may also affect orthostatic CBF. [8]
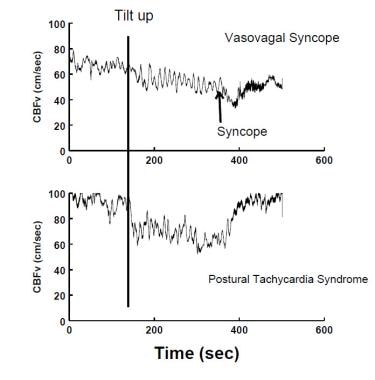 Decreased CBFv measured by transcranial Doppler ultrasound occurs during a VVS (upper panel), and in postural tachycardia syndrome (POTS) (lower panel). During VVS, CBF declines gradually at first and then more abruptly as the patient acutely loses consciousness. In POTS, CBF is fairly uniformly reduced; there is no loss of consciousness although lightheadedness is typical.
Decreased CBFv measured by transcranial Doppler ultrasound occurs during a VVS (upper panel), and in postural tachycardia syndrome (POTS) (lower panel). During VVS, CBF declines gradually at first and then more abruptly as the patient acutely loses consciousness. In POTS, CBF is fairly uniformly reduced; there is no loss of consciousness although lightheadedness is typical.
Orthostatic intolerance is not always due to autonomic or other compensatory dysfunction and can be due to inadequate responses of compensatory mechanisms to environmental stressors. For example, someone who is dehydrated may be unable to stand up without resulting orthostatic hypotension but no autonomic dysfunction is present and therefore neurogenic orthostatic hypotension is not present; instead, the autonomic and other regulatory systems cannot adequately compensate for the loss of circulating blood volume.
On the other hand, neurogenic OI, in which adrenergic vasoconstriction is defective, is associated with primary autonomic failure; patients with related diseases cannot remain standing and have detectable autonomic abnormalities in all postural positions. Therefore, OI encompasses any condition with blood flow, heart rate, and cardiorespiratory regulation inadequacy that are demonstrable in the upright position but may also have abnormal findings in all positions. Under such circumstances, OI is often the most obvious manifestation of a more widespread impairment in integrative neurovascular physiology.
The various types of OI are discussed in the sections that follow.
Initial orthostatic hypotension
Orthostatic hypotension (OH) is defined as a sustained reduction of systolic BP >20 mmHg or diastolic BP >10 mmHg within 3 minutes of standing or following head-up tilt to ≥60o. [9] The requirement of a sustained reduction rules out initial orthostatic hypotension (IOH). This definition is relatively recent and was assembled by a consensus panel in 2011. [9] Before that, there was no consistent definition of OH. Non-neurogenic OH can be caused by drugs, dehydration, blood loss, age, and illnesses that secondarily cause acute or chronic hypovolemia. Neurogenic OH is identified with autonomic failure due to inadequate release of norepinephrine from sympathetic vasomotor neurons leading to vasoconstrictor failure. [9] Neurogenic OH is rare in the young since most causes of autonomic failure are acquired with age either as a primary (e.g., pure autonomic failure, PAF) or secondary (diabetes) disease. Autonomic failure can be primary with pre-ganglionic, post-ganglionic, or both(e.g., Parkinson’s disease) forms of sympathetic dysfunction. [10] However, there exist congenital genetic variants such as Familial Dysautonomia (Riley-Day syndrome) [11] and the exquisitely rare dopamine beta-hydroxylase deficiency (DBH deficiency). [12] Autonomic failure can be autoimmune [13] and may present with the post-infectious Guillain-Barre syndrome although autonomic dysfunction seems to have little effect on ultimate outcome. [14] Autonomic failure is most commonly acquired as a secondary aspect of systemic disease such as diabetes. [15] Sympathetic cardiac denervation is a central aspect of Parkinson’s disease, [16] and may be found in other forms of autonomic failure. Cardiac parasympathetic innervation is also often defective resulting in a steady fall in BP with little reflex tachycardia during orthostatic challenge as shown in the image below.
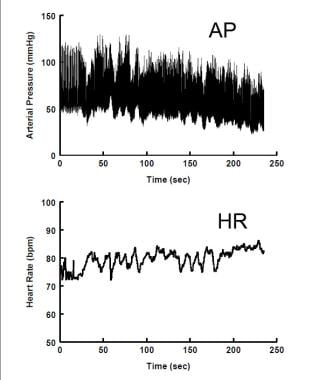 Neurogenic Orthostatic Hypotension. Arterial blood pressure (upper panel) declines steadily during upright stance, while heart rate (lower panel) is only slightly increased.
Neurogenic Orthostatic Hypotension. Arterial blood pressure (upper panel) declines steadily during upright stance, while heart rate (lower panel) is only slightly increased.
Non-neurogenic OH is relatively common in the young. It can be caused by drugs or hypovolemia (e.g., dehydration, hemorrhage). It is by far the most common form of OH in the young. There is no failure of autonomic function but rather incomplete ANS compensation for excessive non-autonomic stressors.
Neurogenic OH (NOH) signifies serious autonomic illness. It is identified with true autonomic vasoconstrictor failure due to the inadequate release of norepinephrine from sympathetic nerves [9] and HR may not increase appropriately with standing.
Postural tachycardia syndrome
Postural tachycardia syndrome (POTS) is defined by day-to-day symptoms of orthostatic intolerance (OI) associated with excessive upright tachycardia but not with hypotension (see image below). [17, 18]
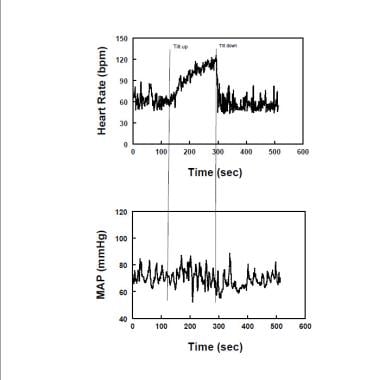 Diagram showing representative heart rate (upper panel) and mean arterial pressure (MAP -lower panel) during upright tilt in a postural tachycardia syndrome patient (POTS). Heart rate increases, while MAP is stable throughout tilt in POTS.
Diagram showing representative heart rate (upper panel) and mean arterial pressure (MAP -lower panel) during upright tilt in a postural tachycardia syndrome patient (POTS). Heart rate increases, while MAP is stable throughout tilt in POTS.
Excessive tachycardia in adults is defined by an upright increase in HR exceeding 30 bpm or to a heart rate exceeding 120 bpm. Recall that the normal HR response to orthostasis is an increase in HR while autonomic failure patients often have no significant increase in HR when upright. Larger heart rate increments are observed in the young with POTS, [19] which is important to know in avoiding over-diagnosis. Symptoms of OI must be concurrent with the excessive tachycardia. No symptoms, no POTS. Tachycardia and concurrent symptoms are very often observed during extremely prolonged orthostatic testing which are therefore to be avoided if the specific diagnosis of POTS is to be made. POTS has often been divided into subgroups designated "neuropathic POTS", in which it is assumed that partial sympathetic denervation or adrenergic hypoactivity is present, and "hyperadrenergic POTS", in which upright adrenergic overactivity dominates the picture. [20]
Neuropathic POTS
As originally described, neuropathic POTS is caused by decreased sympathetic adrenergic vasoconstriction in the lower limbs, associated with reduced leg norepinephrine spillover [21] and reduced vasoconstriction of the lower extremities. [22] There is often increased blood flow ("high flow") in the lower extremities even while supine. Another neuropathic variant has normal lower extremity hemodynamics ("normal flow") but decreased regional sympathetic adrenergic vasoconstriction in the splanchnic circulation. [23] Neuropathic POTS can represent an autoimmune autonomic neuropathy. [13] Thus, when upright, neuropathic POTS patients have greater than normal redistribution of blood to the dependent vasculature causing baroreflex mediated tachycardia and vasoconstriction. The cardiac baroreflex response is also blunted in POTS. [24] Central hypovolemia can also result in hyperpnea and hypocapnia in nearly 50% of patients [7] through a baroreflex mediated mechanism. [25]
Hyperadrenergic POTS
The tachycardia of hyperadrenergic POTS is caused by increased pre-synaptic or post-synaptic adrenergic potentiation. This might include central sympathetic activity and increased sympathetic nerve activity. Increased supine sympathetic activity has been reported, [17] but not universally. [26] To date, our laboratory has only observed increased upright muscle sympathetic activity in one POTS patient. One cause of hyperadrenergic POTS is increased synaptic norepinephrine. The norepinephrine transporter deficiency heterozygote [27] is the prime example of this mechanism but has been found as an autosomal mutation in only one pedigree. Less severe, possibly epigenetic norepinephrine transporter (NET) deficiency has also been demonstrated recently and may have a wider prevalence. [28]
Alternative considerations of mechanism include modulation of the adrenergic synapse through enhanced norepinephrine synthesis and release, and enhanced post-synaptic affinity, which may be modulated by local and humoral transmitters. Thus, for example, the reciprocal actions of nitric oxide (NO) and angiotensin-II respectively reduce and enhance adrenergic activity. The role for NO as an inhibitory neurotransmitter is now well known. [29] Nitrergic NO released from nerves having parasympathetic activity act at pre-synaptic and post-synaptic sites to decrease adrenergic transduction, [30] the process by which a sympathetic nerve impulse causes vascular smooth muscle contraction. This includes reduction of the release and binding of norepinephrine from the neurovascular synapse, [31] interference with post-synaptic neurotransmission, [32] chemical denaturing of norepinephrine, [33] and down-regulation of adrenergic receptors. [34]
Conversely, studies of sympathoexcitation show that angiotensin-II acts through AT1 receptors to increase production of reactive oxygen (ROS) and nitrogen species within the brain at pre-synaptic sympathetic neurons [35] and act in the periphery where they produce pre- and post-synaptic augmentation of sympathetic transduction, and upregulation of adrenergic receptors. [34] In addition, the release and binding of norepinephrine is facilitated, [31] as are the effects of norepinephrine, in the presence of angiotensin-II. This depends critically on the formation of ROS, [36] which also decreases NO, [37] often uncoupling nitric oxide synthase. [38] This mechanism occurs in a variant of "hyperadrenergic POTS" associated with tachycardia, pallor, vasoconstriction ("low flow"), and absolute hypovolemia even while supine. [39] NO, plasma renin, and serum aldosterone are decreased, [40] while plasma angiotensin-II [23] is increased by a defect in ACE-2. [41]
Postural syncope (vasovagal syncope, acute OI, simple faint)
Syncope (fainting) is defined as "complete loss of consciousness due to transient global cerebral hypoperfusion characterized by rapid onset, short duration, and spontaneous complete recovery.” [42, 43] Most syncope is caused by systemic hypotension and reduced cerebral blood flow. It is possible that a cerebrovascular accident could present in similar fashion [44] although it has never been reported in pediatrics. Syncope can be caused by orthostatic hypotension (OH), which has already been discussed. OH is easily ruled out by a 3-minute standing test (see the figure below).
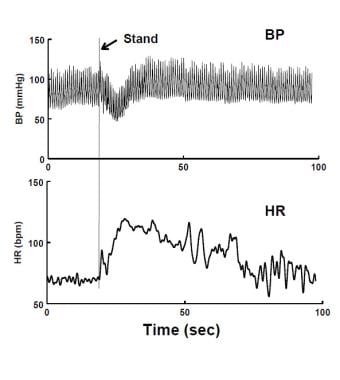 Immediate Orthostatic Hypotension (IOH) upon standing. There is a short-lived decrease in blood pressure (BP - upper panel) and increase in heart rate (HR - lower panel). The fall in BP is resolved within 20 seconds. The patient experienced transient lightheadedness.
Immediate Orthostatic Hypotension (IOH) upon standing. There is a short-lived decrease in blood pressure (BP - upper panel) and increase in heart rate (HR - lower panel). The fall in BP is resolved within 20 seconds. The patient experienced transient lightheadedness.
Syncope is divided among cardiovascular syncope, frequently arrhythmic or structural cardiopulmonary disease, and reflex or neurally mediated syncope. Cardiogenic syncope can be life-threatening and has a poor prognosis unless the cardiac pathophysiology is treated. Cardiogenic syncope is not OI because recumbency does not specifically produce improvement. Reflex syncope has a good prognosis. [45] Reflex syncope includes vasovagal syncope and situational syncope including carotid sinus syncope, [46] which is essentially unknown to pediatrics. Deglutition, [47] defecation, [48] micturition, [49] and cough [50] syncope are rarely observed in the young; and hair grooming [51] and adolescent stretch [52] syncope variants are particular to adolescence. Fainting during exercise raises a "red flag" for cardiogenic syncope and further sport activity should be curtailed until cardiac evaluation is complete. Nevertheless, the most common cause of exercise-related syncope in the young isVVS. [53] Cardiogenic syncope, although not usually posturally related, cannot be automatically dismissed following a first faint. Therefore, the first and consequent episodes prior to cardiovascular evaluation should be treated as urgent. If fainting is subsequently found to be non-cardiogenic then urgency is reduced and simple maneuvers often suffice to acutely deal with the circumstances. Cardiologists are often involved in early evaluation of syncope because initial assessments should determine whether the condition is of cardiac or non-cardiac etiology. Cardiac diseases, when found, are treated specifically. Cardiac syncope may first manifest during exercise, which puts the most physiologic stress on coronary, systemic, and pulmonary circulations and on overall cardiac function. Exercise-related syncope or syncope with cardiac symptoms (e.g., tachycardia, chest pain) indicates a search for underlying heart disease. However, exercise-related syncope in the young is most often noncardiogenic and thephysiology may resemble simple faint.
However, even when neurocardiogenic hypotension is involved, the pathophysiology and clinical history may be more complex, involving changes in respiration with dyspnea and chemoreflex and baroreflex dysfunction that might suggest exercise-provoked heart disease. Still, despite the relative degree of concern attendant to fainting in the young athlete, most such episodes are noncardiogenic in origin. Nevertheless, this should not obviate the need to evaluate such patients for potentially lethal cardiac disease. Cardiogenic syncope is well described elsewhere and is not discussed herein.
Postural syncope and emotional or phobic syncope comprise VVS, the largest subgroup within the reflex syncope category. [54] Regional or system-wide loss of vasodilation is an element in all VVS, at least as a terminal event; it may not always be due to loss of sympathetic nerve activity. Postural syncope is acute OI and approximately two-thirds of patients are female, while teenage boys with this tend to be tall, thin, and rapidly growing. [54] Loss of consciousness is often preceded by a prodrome of OI symptoms, particularly lightheadedness, nausea, sweating, weakness, and blurred vision. Traditionally, postural syncope was believed to be due to reflexes from a hypercontractile, underfilled heart analogous to the Bezold-Jarisch reflex. [55] Evidence to the contrary has accrued; such stimulus would be short-lived. Because of baroreceptor unloading [56] very few afferent nerves were excited in the original experiments by Oberg and Thoren in the moribund hemorrhaged cat. [57] VVS can occur ina ventricular denervated transplant recipient [58] and the heart is neither empty nor hypercontractile prior to syncope. [59] As yet we do not completely understand the pathophysiology of simple faint. [60]
In the most common variant of postural faint occurring in young patients, postural faint comprises three stages (see the image below), which strongly resemble the circulatory changes found during hemorrhage. [61]
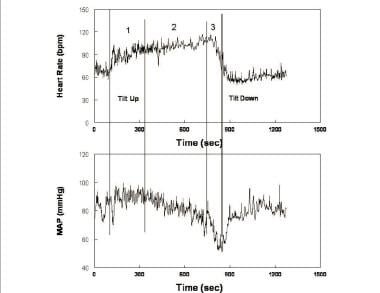 Heart rate (upper panel) and mean arterial pressure (MAP - lower panel) during upright tilt in a representative postural syncope patient. Changes during tilt occur over three stages: during the first stage (1), following initial hypotension, MAP stabilizes slightly higher than resting pressure while heart rate increases. During the second stage (2), MAP begins to fall gradually, while heart rate continues to increase. Note that the increment in heart rate from supine to upright fulfills tachycardia criteria for postural tachycardia syndrome (POTS). During the third stage (3), MAP and then heart rate fall abruptly and rapidly as loss of consciousness supervenes.
Heart rate (upper panel) and mean arterial pressure (MAP - lower panel) during upright tilt in a representative postural syncope patient. Changes during tilt occur over three stages: during the first stage (1), following initial hypotension, MAP stabilizes slightly higher than resting pressure while heart rate increases. During the second stage (2), MAP begins to fall gradually, while heart rate continues to increase. Note that the increment in heart rate from supine to upright fulfills tachycardia criteria for postural tachycardia syndrome (POTS). During the third stage (3), MAP and then heart rate fall abruptly and rapidly as loss of consciousness supervenes.
After initial orthostatic hypotension and restoration of circulatory homeostasis, BP stabilizes while HR increases in Stage 1. BP stability distinguishes postural faint from true OH. BP often exhibits rhythmic fluctuations during this stage referred to as “Mayer waves” [62] with an approximate 10-second period (0.1Hz). Similar periodicity is shared by fluctuations in heart rate, sympathetic nerve activity, and peripheral resistance. The fluctuations are the closed loop time for sympathetic baroreflex response (i.e., the time it takes for detection and compensation for BP changes). [63] Oscillations are accentuated during central blood volume reductions such as occur during orthostasis. During this stage total peripheral resistance increases to sustain BP in the face of a reduced cardiac output (see the image below).
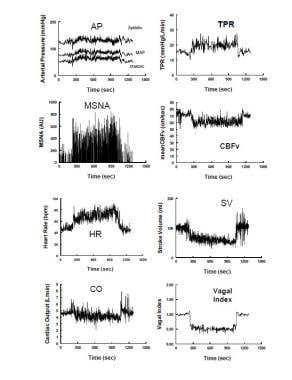 Hemodynamic and neurovascular changes during upright tilt in a representative healthy volunteer. The left panel shows from top to bottom: arterial pressure, muscle sympathetic nerve activity (MSNA) from the peroneal nerve, heart rate (HR) and cardiac output. The right panel shows from top to bottom: total peripheral resistance (TPR), cerebral blood flow velocity (CBFv) by transcranial Doppler ultrasound, stroke volume and a vagal index calculated from the respiratory sinus arrhythmia component of the frequency spectrum of HR variability. During upright tilt at 275 seconds (s), systolic, diastolic and arterial pressures increase slightly, while pulse pressure is decreased with a decrease in stroke volume by approximately 40%. HR increases so that cardiac output is only decreased by 20% because of the increase in HR. CBFv decreases by 5-10%. Both total peripheral vascular resistance and muscle sympathetic nerve activity increase, while the vagal index decreases, reflecting, respectively, sympathetic activation and parasympathetic withdrawal.
Hemodynamic and neurovascular changes during upright tilt in a representative healthy volunteer. The left panel shows from top to bottom: arterial pressure, muscle sympathetic nerve activity (MSNA) from the peroneal nerve, heart rate (HR) and cardiac output. The right panel shows from top to bottom: total peripheral resistance (TPR), cerebral blood flow velocity (CBFv) by transcranial Doppler ultrasound, stroke volume and a vagal index calculated from the respiratory sinus arrhythmia component of the frequency spectrum of HR variability. During upright tilt at 275 seconds (s), systolic, diastolic and arterial pressures increase slightly, while pulse pressure is decreased with a decrease in stroke volume by approximately 40%. HR increases so that cardiac output is only decreased by 20% because of the increase in HR. CBFv decreases by 5-10%. Both total peripheral vascular resistance and muscle sympathetic nerve activity increase, while the vagal index decreases, reflecting, respectively, sympathetic activation and parasympathetic withdrawal.
During Stage 2, BP slowly declines as the baroreflex increases HR further. The decrease in BP is most often related to decreased cardiac output, [46] even though sympathetic activity [64] and peripheral arterial resistance [65] are sustained. Thereafter, resistance and pressure oscillations decrease despite sustained sympathoexcitation. Hyperpnea and hypocapnia occurs at this stage in most patients. [6] In some patients Stage 2 is abbreviated. This is especially true for patients with convulsive syncope in whom episodes occur abruptly in association with asystole.
Convulsive or asystolic syncope (see the image below) is distinguished from epilepsy by decreased EEG activity in the former and by nearly immediate resolution of opisthotonic posturing by recumbence. Despite appearances, asystolic faints are not cardiogenic but reflex mediated and are a relatively uncommon form of simple vasovagal fainting that may also be found in phobic fainting.
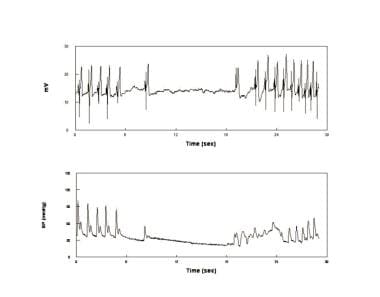 An asystolic faint. Electrocardiogram (upper panel) and blood pressure (BP) (lower panel) in a patient who experienced an asystole during faint. This is episodic, relatively infrequent, and unrelated to intrinsic sinus node disease. Asystolic faints are associated with opisthotonic posturing and have been sometimes referred to as convulsive syncope.
An asystolic faint. Electrocardiogram (upper panel) and blood pressure (BP) (lower panel) in a patient who experienced an asystole during faint. This is episodic, relatively infrequent, and unrelated to intrinsic sinus node disease. Asystolic faints are associated with opisthotonic posturing and have been sometimes referred to as convulsive syncope.
Several mechanisms have been proposed for VVS in some patients. VVS patients with decreased resting BP had reduced tyrosine hydroxylase and NE synthesis, and a group of normotensive had excess NET. [44] A selective deficit of splanchnic vasoconstriction and venoconstriction has also been demonstrated. [22] Prodromal OI symptoms begin during the Stage 2 and clinicians might therefore entertain a diagnosis of POTS in the laboratory setting. Clinical history offers the best way to distinguish patients with acute episodic faints with long periods free of symptoms (postural syncope) from POTS, in which symptoms are chronically present; Indeed, the prodrome of simple faint and the signs and symptoms of POTS are similar because they may have similar initial pathophysiology – excessive reduction in central blood volume resulting in reflex tachycardia. [22, 66, 23] Postural fainters, corresponding to pale and vasoconstricted, hyperadrenergic POTS patients are not generally observed. Forthe most part, in our experience, POTS patients have day-to-day symptoms but do not faint, while syncope patients have episodic faints but not daily symptoms. This distinction has become less clear with time. Thus, some chronic OI (POTS) patients faint, and some episodic fainters also have underlying daily symptoms of OI. However, fainting of POTS patients in laboratory must be viewed cautiously and cannot, by itself, be regarded as proof of real-world fainting. A “real-world” clinical history compatible with fainting is compulsory.
In the last stage, Stage 3, CBF, BP, and HR rapidly fall in that order, seemingly defying BP–CBF causality. [67] Similar effects are often seen in nonlinear systems of all kinds whenever a sufficiently strong external signal entrains linked signals. Thus, recent work shows that both cardiovagal and sympathetic baroreflex efferent arms are impaired prior to fainting, and Mayer waves disappear. Similarly cerebral autoregulation becomes impaired with entrainment of CBF, BP, and HR by an extrinsic signal, which may be hyperpneic respiration. [4, 68] Why baroreflex integrity is lost is not yet known, but this results in abnormal BP-HR and BP-MSNA functional relationships such that HR, BP, and sympathetic nerve activity all decrease resulting in bradycardia, hypotension, and sympathetic silence. [69] The faint is associated with marked systemic vasodilation while CBF falls with declining BP. Recent work challenges the necessity of sympathetic silence as the precipitant of the finalhypotension. [44] While vasodilation always occurs, the sympathetic baroreflex can fail with or without sympathetic silence because of a loss of the functional relationship between blood pressure and sympathetic activation. Loss of functional connections between BP and sympathetic nerve activity, but not heart rate, occurs in patients with vasodepressor syncope where vasodilation without bradycardia occurs. While there is a loss of the sympathetic efferent baroreflex causing progressive loss of compensatory vasoconstriction, the cardiovagal baroreflex remains intact.
POTS and postural syncope are both associated with hyperpneic hyperventilation. [6, 70, 7] Hyperpnea and resultant hypocapnia precede unconsciousness in virtually every vasovagal syncope patient. [6] Hypotension and bradycardia might be explained by the pulmonary stretch reflex unfettered by compensatory baroreflex effects. [71, 68] The cause of hyperpnea is unclear but may relate to the ventilatory efferent arm of the arterial baroreflex. [25] Similar findings of hyperpnea are found in approximately 50% of POTS patients with central hypovolemia who do not faint.
Very frequent or extremely prolonged syncope can point to psychogenic syncope or conversion responses. These are easily distinguished from true syncope in the laboratory because there is no hypotension or reduced CBF, but attacks may be real to the patient. Some patients may have had bonafide VVS interspersed with more frequent psychogenic episodes as learned or conditioned responses. One school of thought suggests that such patients actually experience the symptoms of true VVS without the signs.
Orthostatic intolerance is common but often misunderstood. Investigation of the condition is an evolving field of integrative physiologic study. Postural VVS is identified with acute orthostatic intolerance. Despite its ubiquity, scientists do not yet understand why some people faint. POTS is identified with chronic orthostatic intolerance. POTS, however, remains a heterogeneous entity, likely of varied etiologies. Until better understanding is achieved, treatment remains more guesswork than science.
Pathophysiology
Normal orthostatic regulation
Orthostasis stresses regulatory capabilities of the circulatory system [72] including an intact heart, intact vascular structure and function, adequate blood volume, and intact physical pumps comprising the skeletal muscle pump - leg muscles that compress leg veins - and the respiratory-abdominal muscle pump, which enhances systemic venous return during respiration. [73, 74] Upright stance causes dependent venous pooling. Muscle pumps propel blood back to the heart when upright and during exercise. [73] Enabling the skeletal muscle pump forms an important class of physical “countermeasures” against orthostatic intolerance. [75, 76]
Apart from muscle pumps, rapid orthostatic circulatory adjustments depend on the autonomic nervous system (ANS) comprising sympathetic and parasympathetic arms forming a framework for heart rate (HR) and blood pressure (BP) stability. The myogenic response [77] and flow dependent mechanisms [78] primarily act to ensure tissue level perfusion and autoregulation. The sympathetic arm acts through its primary vascular neurotransmitter norepinephrine [79] , and co-transmitters neuropeptide Y and ATP [80] to produce arterial vasoconstriction and venoconstriction, increase cardiac contractility and HR, stimulate adrenal epinephrine release, and control the neuroendocrine and vascular function of the kidney and long term BP control. The parasympathetic arm via vagal nerve efferents contributes most to heart rate changes at rates less than the intrinsic rate. [81] Recent work indicates strong vagal influences on sympathoexcitation [82] and important effects on nitrergic (nitric oxidecontainingnerves) vasodilation of the large cerebral arteries. [83] Endocrine and local systems (e.g., nitric oxide, local angiotensin) impact the vascular milieu but are slower to develop, often acting to modulate or set tonic activity of the ANS. [80] Autonomic control of HR and BP during orthostasis is provided by subsystems designated “baroreflexes” (pressure reflexes), loosely grouped as arterial and cardiopulmonary baroreflexes, which maintain BP under changing conditions such as orthostasis. [84]
When supine, blood volume within the central thoracic vasculature is relatively large, although a disproportionate amount (25-30%) of blood is stored within the splanchnic venous reservoir. [85] Standing transfers >500ml of central blood caudally, further increasing the volume of the splanchnic pool and filling veins of the lower extremities. [86] An initial period of instability follows, denoted “initial orthostatic hypotension” [87] (see the image below), during which BP can decrease by 30% or more, reaching its nadir at 10-20 seconds after standing. Reflex tachycardia occurs. BP is restored within 30-60 seconds. IOH results from the normal delay of arterial baroreflex detection and response to gravitational blood volume redistribution. Lightheadedness, postural instability, and occasionally brief loss of consciousness occur and can be relieved by recumbency making IOH by far the single most common and innocuous form of orthostatic intolerance. Thereafter, HR decreases butremains elevated compared to supine, and BP is restored by arterial vasoconstriction, by elastic recoil of venous blood in dependent veins, and by active venoconstriction in splanchnic veins. [88]
Immediate Orthostatic Hypotension (IOH) upon standing. There is a short-lived decrease in blood pressure (BP - upper panel) and increase in heart rate (HR - lower panel). The fall in BP is resolved within 20 seconds. The patient experienced transient lightheadedness.
After IOH recovery, sympathetic nerve activity increases while upright blood volume slowly decreases because of microvascular filtration. [89] Decreased venous return decreases central blood volume and cardiac output (CO) by 20% despite baroreflex mediated vasoconstriction, increased cardiac contractility, and increased HR. Cerebral blood flow velocity (CBFv) decreases by 3-12% partly because of reduced cerebral perfusion pressure of 20mmHg. [90] Cerebral autoregulation (unchanged CBF despite changing BP) is blunted during orthostasis. Unless the muscle pump is evoked, standing still places us at risk for decreased CO and CBF. The normal orthostatic response to tilt is shown in the image below.
Hemodynamic and neurovascular changes during upright tilt in a representative healthy volunteer. The left panel shows from top to bottom: arterial pressure, muscle sympathetic nerve activity (MSNA) from the peroneal nerve, heart rate (HR) and cardiac output. The right panel shows from top to bottom: total peripheral resistance (TPR), cerebral blood flow velocity (CBFv) by transcranial Doppler ultrasound, stroke volume and a vagal index calculated from the respiratory sinus arrhythmia component of the frequency spectrum of HR variability. During upright tilt at 275 seconds (s), systolic, diastolic and arterial pressures increase slightly, while pulse pressure is decreased with a decrease in stroke volume by approximately 40%. HR increases so that cardiac output is only decreased by 20% because of the increase in HR. CBFv decreases by 5-10%. Both total peripheral vascular resistance and muscle sympathetic nerve activity increase, while the vagal index decreases, reflecting, respectively, sympathetic activation and parasympathetic withdrawal.
Etiology
Ventricular tachycardia, bradyarrhythmias, and related arrhythmic events are the most common causes of cardiac syncope, but other possible causes include the following:
-
Arrhythmogenic right ventricular dysplasia
-
Cardiomyopathies
-
Left ventricular outflow obstruction
-
Acute or subacute aortic regurgitation (especially postsurgical or endocarditis-related)
-
Primary pulmonary hypertension
Epidemiology
Statistics
Approximately 40% of people will faint during their lives; half of these presenting during adolescence. The peak age for first faint is 15 years old. [91] .
Sex-, age-, and race-related differences in incidence
POTS: Females predominate 3:1, and onset is usually from menarche to menopause.
Non-neurogenic OH is relatively common in the young while neurogenic OH is rare in young people but associated with diabetes, amyloidosis, primary autonomic failure, and Parkinson's disease in older patients. [10] NOH may occur in up to 17% in adults older than 65 years old. [14]
To date, there have been no studies describing the distribution of fainting by racial groups.
Prognosis
Cardiovascular syncope has a poor prognosis unless specifically treated. Reflex syncope has an excellent prognosis. [45] .
Mortality/morbidity
The mortality/morbidity associated with syncope is difficult to evaluate because of its wide range of causes. Patients with cardiogenic syncope accompanying complete heart block, ventricular tachycardia, acute aortic dissection, or pulmonary embolisms are at much greater risk than those with, say, vasovagal syncope. Appropriate clinical evaluations of the cause of syncope are therefore necessary and appropriate.
Patient Education
In many instances, patients with postural syncope experience a prodrome of symptoms. When these occur, patients should be instructed to employ so-called physical countermaneuvers to obviate postural effects. These include the use of bilateral handgrip for 15 seconds before rising as this forestalls IOH by evoking the exercise pressor reflex, which can substantially increase blood pressure. Lower body muscle contraction while seated (e.g., pumping calf muscles) can also help as a preemptive maneuver. Also, lower body muscle tensing of legs, buttocks, and abdomen particularly attenuates the transient arterial blood pressure decrease once standing has occurred and can be potentiated by handgrip. Recumbence and squatting are general measures used to remediate all forms of orthostatic intolerance. Recommendations for patients to increase salt and water intake may also be of help.
-
Decreased CBFv measured by transcranial Doppler ultrasound occurs during a VVS (upper panel), and in postural tachycardia syndrome (POTS) (lower panel). During VVS, CBF declines gradually at first and then more abruptly as the patient acutely loses consciousness. In POTS, CBF is fairly uniformly reduced; there is no loss of consciousness although lightheadedness is typical.
-
Neurogenic Orthostatic Hypotension. Arterial blood pressure (upper panel) declines steadily during upright stance, while heart rate (lower panel) is only slightly increased.
-
Diagram showing representative heart rate (upper panel) and mean arterial pressure (MAP -lower panel) during upright tilt in a postural tachycardia syndrome patient (POTS). Heart rate increases, while MAP is stable throughout tilt in POTS.
-
Immediate Orthostatic Hypotension (IOH) upon standing. There is a short-lived decrease in blood pressure (BP - upper panel) and increase in heart rate (HR - lower panel). The fall in BP is resolved within 20 seconds. The patient experienced transient lightheadedness.
-
Heart rate (upper panel) and mean arterial pressure (MAP - lower panel) during upright tilt in a representative postural syncope patient. Changes during tilt occur over three stages: during the first stage (1), following initial hypotension, MAP stabilizes slightly higher than resting pressure while heart rate increases. During the second stage (2), MAP begins to fall gradually, while heart rate continues to increase. Note that the increment in heart rate from supine to upright fulfills tachycardia criteria for postural tachycardia syndrome (POTS). During the third stage (3), MAP and then heart rate fall abruptly and rapidly as loss of consciousness supervenes.
-
Hemodynamic and neurovascular changes during upright tilt in a representative healthy volunteer. The left panel shows from top to bottom: arterial pressure, muscle sympathetic nerve activity (MSNA) from the peroneal nerve, heart rate (HR) and cardiac output. The right panel shows from top to bottom: total peripheral resistance (TPR), cerebral blood flow velocity (CBFv) by transcranial Doppler ultrasound, stroke volume and a vagal index calculated from the respiratory sinus arrhythmia component of the frequency spectrum of HR variability. During upright tilt at 275 seconds (s), systolic, diastolic and arterial pressures increase slightly, while pulse pressure is decreased with a decrease in stroke volume by approximately 40%. HR increases so that cardiac output is only decreased by 20% because of the increase in HR. CBFv decreases by 5-10%. Both total peripheral vascular resistance and muscle sympathetic nerve activity increase, while the vagal index decreases, reflecting, respectively, sympathetic activation and parasympathetic withdrawal.
-
An asystolic faint. Electrocardiogram (upper panel) and blood pressure (BP) (lower panel) in a patient who experienced an asystole during faint. This is episodic, relatively infrequent, and unrelated to intrinsic sinus node disease. Asystolic faints are associated with opisthotonic posturing and have been sometimes referred to as convulsive syncope.





