Practice Essentials
Posttransplant lymphoproliferative disease (PTLD) is an unusual entity that has many features of an immune system malignancy. It is characterized by uncontrolled proliferation of lymphoid lineage cells (typically B cells, rarely non-B lineage) in a context of posttransplant immunosuppression. [1, 2] In some situations, reducing the immunosuppression can reverse this proliferation, thus differentiating PTLD somewhat from truly irreversible malignancies. Most but not all PTLD cases have a strong relationship with Epstein-Barr virus. [3, 4, 5] This condition straddles the disciplines of transplantation, immunology, oncology, and virology.
PTLD has emerged as a significant complication of solid organ transplantation. [6] This entity is difficult to predict or prevent and has high morbidity and mortality rates. In addition, it poses the potential for graft loss due to the disease itself or the need to reduce immunosuppression, which increases the risk of graft rejection.
Incidence
Multiple reports from single-center studies, as well as registry reports, have illustrated that the incidence of PTLD rose significantly in the 1990s. [7, 8] For example, the North American Pediatric Renal Transplant Cooperative Study (NAPRTCS) showed a doubling of incidence density (cases per 100,000 years of patient follow-up) from 320 in 1987-1992 to 630 in1992-1997. [8] Since the turn of the century, the incidence has been decreasing. [9, 10]
Some centers, particularly those performing pediatric liver transplantation, introduced prophylactic measures and reported a reduction in the high rate of PTLD at their institutions. [11, 12] Current prevalence rates for posttransplant patients vary from 1-15%, depending on the organ transplanted and the immunosuppressive agents used. [13]
Notably, in studies of newer biologic agents such as belatacept and tofacitinib the incidence of PTLD has been higher in treatment than control groups. [14] An expert multidisciplinary panel felt that the higher incidence might also reflect the increasing use of transplantation in higher-risk patients who need intense immunosuppression, such as those with preformed high-level antibodies. [15]
Risk Factors
The prevalence of PTLD is different for each transplanted organ. The highest rates are reported for the intestine (as much as 20%), thoracic organs (heart 2-10%, lung 4-8%), and liver (2-8%). Prevalence in kidney transplants is usually lower (1%), but some centers have reported prevalence as high as 10%. Prevalence in bone marrow transplantation is low (1-2%), except in patients in whom T-cell–depleted marrow is used; in these patients, rates as high as 24% are reported.
In an analysis of the very large national United Network for Organ Sharing (UNOS) registry in the United States, the prevalence of PTLD was highest in intestinal transplants (8%), then thoracic organs (3-5%), followed by liver and kidney transplants. [16] In addition, several different demographic, infectious, and immunosuppressive risk factors are associated with PTLD development.
Age- and race-related risk
The prevalence of PTLD is highest in the pediatric age group (age 0-18 y). According to analysis of UNOS data, the relative risk (RR) in this age group is 2.81 compared with adult recipients. [16] Some reports have suggested that prevalence is even higher in patients younger than 5 years.
Age may not be an independent risk factor but may depend on the likelihood of the recipient being seronegative for Epstein-Barr virus (EBV) at the time of transplant. In general, among transplant recipients, the risk of lymphoma (a subgroup of PTLD) is highest in youngest patients, ranging from 30-fold in the Australia and New Zealand Dialysis and Transplant Registry (ANZDATA) kidney transplant database and 200- to 1200-fold in the 42-country Collaborative Transplant Study (CTS) registry. [17] However, even adult recipients, especially older adults, are at a higher risk than the general population (7- to 16-fold in both registries). [17, 18]
Data from the NAPRTCS and UNOS registries have shown a markedly higher risk of PTLD in white children; the relative risk for white transplant recipients is 2.22 compared with that in other races. [16]
Infectious risk factors
The single most important risk factor for PTLD is the lack of previous exposure to EBV in the transplant recipient. Across all recipient age groups and all organ types, recipient EBV seronegativity confers a 3- to 33-fold higher risk for PTLD. [19] This magnitude of elevation is by far higher than with any other factor. Walker et al determined from their data on 381 adult non-kidney transplant recipients, 14 of whom developed PTLD, that the prevalence rate for seronegative recipients was 24 times higher than that for seropositive recipients. [20] Gain of function mutations in the Latent Membrane Protein 1 at G212S and S366T carried a nearly 12-fold increased risk of development of EBV-positive PTLD. [21]
Cytomegalovirus (CMV) infection has also been shown to be associated with an increased risk of PTLD. Manez et al analyzed 40 adult liver transplant recipients, all of whom were seronegative for EBV. [22] Of these recipients, 33% developed PTLD, and a diagnosis of CMV disease posttransplant increased the relative risk to 7.3. In a study in adult nonrenal transplant recipients by Walker et al, CMV-seronegative recipiens who received an allograft from a CMV-positive donor were at 4- to 6-fold higher risk of PTLD. [20]
Immunosuppression risk factors
PTLD was extremely rare when two-drug immunosuppression regimens (steroids and azathioprine) were in standard use. The incidence of PTLD rose with the advent of more potent immunosuppression, beginning with cyclosporine. In one long-term study, the relative risk for PTLD with cyclosporine was significantly higher at 2.2. [23] In addition, certain specific agents have been reported to increase the risk of PTLD.
Swinnen et al first reported the increased prevalence of PTLD (more than 4-fold) in recipients of cardiac transplants who received more than a 10-mg cumulative dose of muromonab-Cd3 (OKT3). [24] Walker et al reported that the use of OKT3 independently increased the risk of PTLD 5- to 6-fold in a multivariate analysis. [20] In combination with the risk factors of an EBV-seronegative recipient and CMV, the prevalence rate rose by a factor of 529.
Cox et al and Sokal et al reported that pediatric recipients of liver transplants who received FK506 (tacrolimus) had a higher prevalence of PTLD (13-20%) than those who received cyclosporine (2-3%). [25, 26] According to Newell et al, the combined use of OKT3 and FK506 appeared to be synergistic, with the prevalence of PTLD increasing from 6% to 28%. [27] Initial data from NAPRTCS also suggested a marked increase in PTLD with FK506-based initial immunosuppression for kidney transplantation. [8] However, more recent data suggest no increased risk. This may be related to lower levels of tacrolimus being deemed acceptable (8-12 ng/mL, compared with 12-15 ng/mL previously).
Penn has commented that each drug used is associated with a learning curve, during which time increased adverse effects may be observed. [28, 29] Whether rates of PTLD will decrease in liver transplantation with lower tacrolimus levels is unclear. The Cochrane Review Group compared cyclosporine with tacrolimus and found no significant differences in relative risk for PTLD in a meta-analysis of 30 different prospective trials. [30, 31]
Studies that have examined the risk for PTLD with mycophenolate mofetil have not shown any increase in relative risk; these studies included a UNOS data analysis, [32] a case control study, [33] and a NAPRTCS registry data analysis. [34] However, other studies have documented an increased risk for BK virus nephropathy and CMV infection with mycophenolate use.
Sirolimus has unique properties, including retention of anti-EBV T-cell activity in vitro. A study of UNOS data revealed a decreased relative risk for malignancy (a different outcome variable from PTLD) with sirolimus use. [32] However, more recent data from the same registry suggested a higher PTLD risk with early sirolimus use, so the issue may not be as clear-cut as thought. [35]
Data regarding PTLD risk with use of anti-interleukin (IL)-2R antibodies (basiliximab and daclizumab [no longer available in the US) have shown conflicting results. Bustami et al analyzed data from the Scientific Registry of Transplant Recipients (SRTR) and documented significantly increased relative risk (1.83-1.92) for PTLD with these agents. [36]
However, Cherikh et al, using similar source data from UNOS, suggested an insignificant 14% increase in relative risk. [32] Opelz and Henderson, who represented the Collaborative Transplant Study in Europe, found no increased risk for non-Hodgkin lymphoma within a 12-month follow-up period after anti–IL-2R antibody use. [37] These differences may be related to different databases and different time periods of follow-up.
To date, alemtuzumab has not been associated with a higher PTLD risk. [35] However, belatacept, a new co-stimulatory blockade agent, is associated with higher PTLD rates, especially in recipients who were EBV seronegative at transplantation. [14]
Other
Some single-center studies have shown associations with individual HLA alleles, such as HLA-A2, -A3, -A8, or -A26. [38] However, none of those allele associations has yet been reproduced by others.
While PTLDs are a heterogeneous group, some separation may be possible between early- and late-onset PTLDs. Early-onset PTLDs tend to be EBV positive and, when extranodal, are more likely than late-onset PTLDs to be localized to the transplanted organ. Late-onset PTLD is less likely to be associated with EBV and, overall, is more likely than early-onset PTLD to be extranodal. [39]
In a French study, simultaneous kidney-pancreas transplantation and higher degree of HLA mismatch (5-6 versus 0-4) imparted higher risk. [13]
Etiology and Pathogenesis
The pathogenesis of PTLD is intimately linked to EBV. [40] EBV is a lymphotrophic DNA gamma herpes virus that replicates in squamous epithelial cells of the oropharynx, uterine cervix, and male genital tract. The virus infects and immortalizes B lymphocytes that bear EBV membrane C3d receptors.
The free infectious virus can be recovered from saliva in essentially all healthy seropositive individuals. EBV is implicated in the pathogenesis of a spectrum of B-cell lymphoproliferative diseases in immunosuppressed organ transplant recipients, in immunodeficiency diseases (eg, common variable immunodeficiency, Wiskott-Aldrich syndrome, ataxia telangiectasia, severe combined immunodeficiency, acquired immunodeficiency syndrome), and in recipients of T-cell–depleted or mismatched bone marrow transplants.
EBV causes 2 types of cellular infections: (1) a productive replicative infection in which mature infectious virus particles are assembled and released, resulting in cell death (the lytic cycle), and (2) a nonproductive infection in which the virus is incorporated into and replicates with the host DNA but remains in the latent state in transformed B cells and no mature virus is produced.
Persistence of the EBV genome in the latent state in transformed B cells follows primary EBV infection and results in a permanent carrier state, in which small numbers of latently infected B cells circulate in seropositive individuals. Elimination of these cells is carried out by human leukocyte antigen (HLA)-restricted, EBV-specific, cytotoxic T lymphocytes. Certain factors (eg, inhibition of anti-EBV T-cell immunity, such as that which occurs with posttransplant immunosuppression) allow latently infected cells to enter the lytic cycle.
Carriage of certain HLA-E alleles has been shown to affect host response to EBV. Individuals with the HLA-E∗0103/0103 genotype are less likely to experience infectious mononucleosis from primary EBV infection, compared with those with the HLA-E∗0101/0101 genotype, but are significantly more likely to develop EBV-positive PTLD. In contrast, transplant recipients with symptomatic EBV reactivations but without PTLD were significantly more likely to have the HLA-E∗0101/0101 genotype. [41]
The suppression of EBV-specific CD8+ T cells also allows B-cell proliferation to go unchecked. In solid organ transplant recipients, the abnormal B cells are usually of recipient origin. In contrast, the abnormal B cells are usually of donor origin in recipients of bone marrow transplants.
According to the above paradigm, all PTLDs should represent B-cell proliferations secondary to EBV infection; however, T-cell and natural killer (NK)–cell PTLDs have also been reported. Penn estimated that 87% of all PTLDs were of B-cell origin, 13% were of T-cell origin, and 0.5% were of null cell origin. [29]
PTLDs not associated with EBV have also been reported. A higher proportion of late-developing PTLDs (> 2 y posttransplant) are more likely to be non–B-cell related or non-EBV related. Thus, gaps are still recognized in the understanding of the pathogenesis of PTLD.
Although EBV can express up to 100 genes, in the posttransplant situation only 9-10 genes are expressed. The EBV genome adopts an episomal configuration and expresses proteins such as BCRF1, BARF1 and viral IL-10 that help avoid immune detection, and some in vitro studies suggest that EBV genes can integrate into the host genome. [9]
The latent membrane proteins LMP1 and LMP2 are believed to act as oncogenes, allowing B cells to escape cell death and proliferate in uncontrolled fashion. However, in a study by Vietzen et al, only certain LMP1 variants were associated with risk for PTLD. [41]
In one study, some PTLDs demonstrated mutations in bcl-6, an intracellular protein of the bcl group of proteins that are involved in passive cell death pathways. Polymorphisms in 2 key anti-inflammatory cytokines, IL-10 and tumor growth factor (TGF)-beta, are associated with susceptibility to EBV-associated PTLD, suggesting that a shift in pro-inflammatory/anti-inflammatory response is involved in the pathogenesis of PTLD. [42] Expression of latency III genes and XBP1 gene are associated with worse outcomes. [43]
Classification of posttransplant lymphoproliferative disease
Two major different classification schemes for PTLD were previously proposed to compare outcomes and determine prognosis: the World Health Organization (WHO) classification and the Harris classification. [44] Both schemes are based on the following characteristics:
-
Clinical
-
Histologic
-
Immunologic cell typing
-
Cytogenetic
-
Immunoglobulin gene-rearrangement
-
Virologic
The classification schemes have common features, including benign hyperplasia or mononucleosis as the mildest form, characterized by maintenance of the nodal architecture; malignant lymphoma, with all the features of malignancy, as the most severe form; and polymorphic or polyclonal proliferations (with nodal architecture destruction and local invasion) classified in the intermediate categories.
Neither system addressed the full range of clinical, virologic, immunologic, and disease markers, and they related poorly to disease prognosis and treatment. A third system, the Ann Arbor staging system, though applied to PTLD, was developed for Hodgkin lymphoma and has limitations in situations with localized intragraft disease, which actually has a good prognosis.
An expert multidisciplinary group met in Seville in 2009 and proposed a new classification that adds important clinical and virologic features of PTLD to the 2008 WHO histologic classification system, making the schema more mechanistic and possibly more prognostic. [15] The subsequent 2016 WHO classification system recognizes 6 major histopathologic subtypes of PTLD, as follows [45] :
-
Plasmacytic hyperplasia PTLD
-
Infectious mononucleosis PTLD
-
Florid follicular hyperplasia PTLD
-
Polymorphic PTLD
-
Monomorphic PTLD (B- and T-/NK-cell types)
-
Classic Hodgkin lymphoma PTLD
Depending on the interplay of immunosuppressive effect and B-cell proliferation, patients may develop uncomplicated mononucleosis or polyclonal polymorphic B-cell hyperplasia, both of which depend on continued viral replication. These benign PTLDs can spontaneously resolve (if host immune response to the virus is adequate) and/or respond to antiviral therapy that interrupts the EBV replicative cycle.
In some patients, these benign PTLDs may progress to an intermediate stage in which a small subpopulation of malignantly transformed cells is present in a predominantly polyclonal proliferation. This second step may involve a cytogenetic event or selection that confers malignant growth potential on an EBV-infected B cell, thus leading to the outgrowth of a malignant cell or single clone analogous to the pathogenesis of African Burkitt lymphoma.
In other patients, the malignant cell clone may become the dominant proliferating cell type, leading to frank lymphoma. Tsao et al have provided a review of the pathologic classifications and evolutions. [46]
Much remains to be understood about the factors involved in determining the severity of PTLD in an individual patient.
Clinical Features
Clinical features of PTLD can be multiple, varied, and complex. In many patients, the early symptoms are nonspecific, including fever, malaise, and weight loss. Maintaining a high index of suspicion for PTLD in all transplant recipients is strongly recommended.
The most common presentation is with sudden-onset lymphoid mass swelling, either externally (eg, cervical lymph nodes) or internally (eg, abdominal or intracranial masses). Extranodal tumors are more common than nodal tumors. An estimated 20-25% of these masses develop within the graft, such as the kidney or liver, and present as allograft dysfunction. [47]
Symptoms relate to the secondary effects of the tumor, such as abdominal pain, respiratory difficulty, stridor, and seizures. CNS presentation is associated with a poorer prognosis. Extra-abdominal lymph node localization occurs in 10-33% and abdominal lymph node or GI tract localization in 10-29% of cases. [17, 19] CNS localization is rarer, seen in 9-13% of cases, except in the trials of belatacept, in which CNS localization was more frequent.
PTLD may present as a fever of unknown origin in the transplant recipient or may mimic graft rejection, particularly late rejection.
The time to PTLD diagnosis posttransplant can widely vary, ranging from a few months to several years. The mean time in most series is 20-35 months, but this is skewed by the long-range interval. The median times are much shorter, approximately 4-5 months. NAPRTCS data and a report by Alfrey et al show a reduction in median time to PTLD at the end of the 20th century. [8, 48]
Diagnosis
The diagnosis of PTLD may often be complicated. Initial studies are often focused on the manifesting symptoms, such as imaging localization of the mass and determination of its extent, invasiveness, and homogenicity. Ultrasonography, CT scanning, or MRI can be used, depending on the area of interest.
Pickhardt et al have published an extensive series of reviews of imaging features of PTLD by body segment. [49, 50, 51, 52, 53] CT scanning is preferred for abdominal and chest imaging. Either CT scanning or MRI can be performed for neuroimaging. Bone marrow biopsies are necessary to help define marrow involvement, which may rarely be the only affected tissue. These imaging and biopsy tests may also be repeated after treatment to assess the response.
Concomitant serologic tests (ie, immunoglobulin [Ig] G and IgM) should be performed to determine recent EBV or CMV primary or secondary infection. The sera can also be analyzed for Epstein-Barr–viral capsid antigen (EB-VCA).
Histopathologic diagnosis of biopsy tissue remains the criterion standard for making the diagnosis of PTLD. Using light microscopy, infiltrates of polymorphous or monomorphous mononuclear cells (small lymphocytes and plasma cells) are observed that disrupt the architecture of the invaded tissue, such as the lymph node. Depending on the degree of proliferation and dedifferentiation, lesions may be characterized as hyperplastic, lymphomatous, or intermediate.
Abnormal cells may be positive for the B-cell markers CD19, CD20, CD21, or CD22, although not in every patient. Lymphocyte CD20 expression is of value in determining therapy choices. Determination of cell surface heavy and light chain Ig expression allows for classification as polyclonal or monoclonal. Detailed cytogenetic studies may be needed in patients with nuclear changes suggestive of frank malignancy.
EBV can frequently be revealed within the abnormal cells using various methods. Epstein-Barr–encoded RNA can be detected by the Epstein-Barr early region (EBER) immunostaining assay (see the image below). The EBV latent membrane protein (LMP) can also be identified through immunostaining. Some PTLDs demonstrate Reed-Sternberg–like cells that may cause confusion with Hodgkin lymphoma; CD15 staining is typically positive in true Hodgkin lymphoma and negative in the Hodgkin lymphoma–like condition.
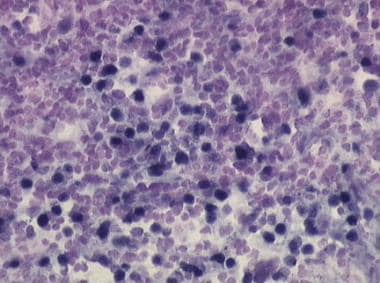 Epstein-Barr viral RNA can be detected by Epstein-Barr early region (EBER) assay (dark-blue staining cells) in the abnormal B cells from tumor masses of posttransplant lymphoproliferative disease (PTLD).
Epstein-Barr viral RNA can be detected by Epstein-Barr early region (EBER) assay (dark-blue staining cells) in the abnormal B cells from tumor masses of posttransplant lymphoproliferative disease (PTLD).
PTLD may occur alongside acute rejection, and the diagnoses may be difficult to separate, especially with T-cell PTLD within the allograft. According to Nalesnik, the following features favor the diagnosis of PTLD in such cases [54] :
-
Nodular infiltrates
-
Serpiginous necrosis
-
Plasmacytoid and immunoblastic cells
-
Absence of ancillary cells such as neutrophils
In an effort to improve the likelihood of successful therapeutic interventions, many investigators have studied methods that may result in early identification of disease in patients at high risk for this disorder. Monoclonal proteins, particularly IgM-related proteins, have been reported to appear with greater frequency in serum and urine of patients with PTLD (71%) than in patients without PTLD (27%). Darenkov et al reported that CD19+ B cells could be detected in the peripheral blood of patients with PTLD but not in those without PTLD, provided no antiviral prophylaxis was used. [55]
Of greater promise are the variations of quantitative or semiquantitative polymerase chain reaction (PCR) techniques to detect EBV viral DNA from the peripheral blood lymphocytes. These techniques measure DNA in blood that can be membrane bound or free. Multiple studies have demonstrated that increased EBV viral loads can be used to differentiate between latent infection and PTLD. Rooney et al showed that levels of EBV DNA between 20,000 and 200,000 copies per microgram of peripheral blood DNA were associated with subsequent PTLD (normal < 2000 copies/μg). [56]
In a study by Rowe et al, an EBV genomic load greater than 500 copies per 100,000 lymphocytes was associated with PTLD development. [57] Wadowsky et al reported that whole blood or peripheral blood mononuclear amplification by competitive reverse transcriptase–PCR gives comparable results but amplification from plasma does not. However, different techniques in different labs and different cut-off values between labs prevent standardization of results between those labs.
In pediatric patients, viral load monitoring is associated with high sensitivity but poor specificity. Many pediatric patients exhibit higher viral loads than adults because of a primary EBV infection or a chronic high viral load state without ever developing PTLD. [58] In anecdotal cases, pediatric PTLD lesions that were EBER or LMP stain positive were not associated with a high viral load in peripheral blood. [59] In contrast, Tsai et al reported that in adult transplant recipients, EBV viral load monitoring was associated with a high specificity but low sensitivity, the opposite of what has been reported in children. [60] This may be related to a relatively higher prevalence of non-EBV, T-cell type, or late-onset PTLD in adult patients.
Rooney et al reported that, in patients who had received bone marrow transplants, spontaneous outgrowth ex vivo of B cells transformed with EBV from the peripheral blood was associated with PTLD to a very high degree of sensitivity and specificity. [56]
DNA levels may also provide evidence of successful therapy. A drop in viral load after intervention suggests a good response. [61] However, this may not hold true after anti-CD20 antibody treatment, where viral loads may remain high because of release of viral DNA from lysed B cells.
Differential diagnosis
A high index of suspicion for PTLD must be maintained in all transplant recipients. Dharnidharka et al reported several cases of catscratch disease that manifested with fever and lymphadenopathy and resembled PTLD. [34] The diagnosis was suspected after inquiry for cat exposures and was confirmed by (1) serologic detection of IgM antibodies to Bartonella henselae and (2) detection of Bartonella species in the lesions by Steiner stain. This condition is often self-limiting in immunocompetent patients and amenable to antimicrobial therapy (sulfamethoxazole-trimethoprim, gentamicin) in immunosuppressed patients. In developing countries, tuberculosis is endemic and also manifests as fever and lymphadenopathy.
Treatment and Prophylaxis
Treatment
No uniform consensus regarding optimal treatment options for PTLD is available. This largely reflects the current multiple gaps in knowledge of this disorder. Guidelines from the American Society of Transplantation Infectious Diseases Community of Practice note that reduction in immunosuppression is the best-validated intervention. For treatment of CD20+ PTLD, the guidelines recommend response-dependent sequential use of reduction in immunosuppression, rituximab, and cytotoxic chemotherapy. [21]
Most centers recommend a staged treatment regimen based on the degree of clonality and aggressiveness. Treatment recommendations may rapidly change with newer understanding of the disease pathogenesis. [62]
Reduction of immunosuppression is the first intervention recommended in most cases and was effective in an early PTLD series by Starzl et al (1984). [63] Most centers reduce the dose of calcineurin inhibitors, and many centers discontinue calcineurin inhibitors entirely if the disease is severe. Many centers also discontinue or reduce mycophenolate or azathioprine. Corticosteroids are usually continued without dosage modification.
In mild cases, reduction of immunosuppression may be the only intervention required. Graft rejection is an obvious risk but, surprisingly, is not observed in every patient. Immunomodulation by the underlying disease is speculated to prevent the normal alloimmune response in these patients.
In a single single-center retrospective review of prospectively enrolled patients, treatment was tailored to standard-risk patients, who received immunosuppression reduction and rituximab, or high-risk patients, who received immunosuppression discontinuation, rituximab and polychemotherapy. The cumulative incidence of rejection at 1 and 5 years after the diagnosis of PTLD was 35% (95% CI, 18-69%) and 53% (33-85%), respectively, whereas the disease-free survival at 1 and 5 years was 94% (95% CI, 65-99%) and 75% (45-90%), respectively. [64]
Anti-CD20 monoclonal antibody has been used, with promising results, to neutralize the CD20-expressing B cells. Several small uncontrolled series have documented good success rates (complete or partial remission) in the 60-70% range in polymorphic or polyclonal cases. In some situations, the PTLD lesions may not express CD20; thus, rituximab may not be useful in such cases. This agent has become the second-line therapy at many centers, overtaking the use of interferon-alfa. Some investigators have proposed its use as first-line therapy, but no studies have directly compared rituximab versus reduction of immunosuppression.
Interferon-alfa is also reported to be efficacious in treatment of PTLD that is unresponsive to immunosuppressive dose reduction alone. Some protocols combine this agent with intravenous IG (IVIG) or CMV-specific Ig. Because of a higher prevalence of acute rejection episodes after interferon use, this agent is less commonly used.
The role of antiviral drugs in treatment of PTLD is controversial. Acyclovir and ganciclovir are effective against replicating virus but are reportedly not effective against latent EBV, which is the predominant form of EBV in PTLD. However, a few virions entering the lytic cycle are possible, and antiviral agents may eliminate these virions. Many centers thus add antiviral therapy to their regimens to eliminate any replicating virus that may be present. The expression of Z EBV replication activator (ZEBRA) in PTLD tumors suggests lytic cycle replication is ongoing. [65]
Standard cancer chemotherapy is reserved for patients with definitive features of malignancy. The usual regimen used has been the CHOP (cyclophosphamide, doxorubicin [hydroxydaunorubicin], vincristine (Oncovin), prednisone) regimen used for non-Hodgkin lymphoma. Gross et al used a modified CHOP protocol with lower doses of cyclophosphamide to prevent toxicity. [66] More recently, Trappe et al showed that sequential use of chemotherapy when rituximab has failed (and vice versa) can be effective. [67, 68]
A sequential approach to immunosuppression reduction, followed by rituximab and CHOP chemotherapy, preserved the glomerular filtration rate (GFR) in kidney transplant recipients with PTLD. [69] This regimen has shown consistently good results in a larger international trial. [70]
Another possible chemotherapy regimen that allowed for total discontinuation of immunosuppression in a small pediatric series incorporated fludarabine, cyclophosphamide, doxorubicin, and rituximab. [71]
Newer agents that have been tried in non-Hodgkin lymphoma but not tried yet in PTLD include radio-conjugated anti-CD20 antibodies such as tositumomab (anti-CD20 tagged to I131) and ibritumomab (anti-CD20 tagged to Y90). Resveratrol has shown some promise in in vitro models. [72] The checkpoint inhibitor nivolumab has been reported as a treatment option. [73] In CNS PTLD, a combination of drugs, including zidovudine, rituximab, and ganciclovir, has shown sustained remission in one study. [74]
Prognosis
The prognosis of PTLD widely varies. Most mild cases regress, but graft rejection may occur (reported rates vary widely from 10-60%).
The outlook for more severe cases is less favorable, particularly in frank malignancies or CNS PTLD. Although individual centers with high PTLD rates report good patient and graft survival rates, multicenter registry data suggest somewhat poorer graft survival outcomes. Non-EBV–positive or late-onset cases have a poorer prognosis. Children generally have a better prognosis. More recent cases of PTLD have a better prognosis than those from older eras. [75] A prognostic score specific to PTLD has been developed in France. [76] PTLD tissue positivity for anelloviruses by metagenomic sequencing or PCR was associated with higher post-PTLD mortality [77] .
In cases in which graft loss occurs, retransplantation has been successfully performed. Johnson et al published a large national registry series of retransplants after PTLD. [78] PTLD recurrence in the repeat transplant has not been reported.
Prophylaxis
With the role of EBV and CMV infections in the development of PTLD established, attention turned toward prevention of these infections posttransplant in an effort to reduce prevalence of PTLD. [79] Prevention can be primary or secondary.
Primary prevention includes the following:
-
Antiviral vaccine
-
Immunization minimization to prevent infection
-
Immunoprophylaxis via intravenous immunoglobulin preparations
-
Chemoprophylaxis via antiviral drugs
Immunization
The development of a vaccine to prevent EBV infection has been a research goal for decades. Several candidate vaccines have been tested in preclinical trials and a few have entered phase 1/2 clinical trials, but none have yet to progress to a phase 3 clinical trial. Much of the vaccine research has focused on gp350, a glycoprotein involved in viral entry into host cells; more recently, a multivalent approach that combines gp350 with other EBV envelope glycoproteins (eg, gH/gL, gB) has shown promise. [80, 81]
Vaccines being studied for treatment of EBV infection consist of platforms that express the EBV latent proteins EBNA1, LMP1, and/or LMP2. These therapeutic vaccines have been shown to promote specific CD4+ and CD8+ cytotoxic responses, with antitumor activity. Adding EBV envelope glycoproteins may increase the efficacy of therapeutic EBV vaccines. [80]
Chemoprophylaxis and immunoprophylaxis
Ganciclovir and acyclovir are antiviral agents that have demonstrated efficacy against EBV and CMV. Davis et al reported in a retrospective analysis that the prevalence of PTLD appeared to be lower with the use of concomitant antiviral therapy (ie, initially IV ganciclovir, then oral acyclovir) in adult transplant recipients. [82]
Darenkov et al conducted a prospective trial of prophylactic antiviral agent use during antilymphocyte antibody administration to adult transplant patients. [55] Using acyclovir if both donor and recipient were CMV seronegative or ganciclovir if either were seropositive, prevalence of PTLD dropped to 0.5% in their study group, compared with 3.9% in historical controls.
However, Green et al were unable to document any change in PTLD prevalence in a prospective trial utilizing acyclovir. [83] In their study, initial intravenous ganciclovir followed by prolonged oral acyclovir was associated with a trend towards increased PTLD and significantly higher CMV incidence. Similarly, CMV-Ig did not have any impact on EBV infection rate or PTLD rate. [84] In contrast, a large 2007 retrospective registry analysis by Opelz et al suggested a significant benefit to IVIG or CMV-specific Ig. [85]
Funch et al performed a large retrospective case-control analysis that showed that ganciclovir was associated with a significant reduction in the odds ratio for PTLD (0.62; 95% CI, 0.38-1.0). [86] Acyclovir was associated with a lower and nonsignificant reduction. The addition of IVIG to ganciclovir did not further reduce the PTLD rate. [87]
The newer oral agents, valacyclovir and valganciclovir, have greater bioavailability but have not been tested for PTLD prevalence reduction, although many centers now routinely use valganciclovir for prophylaxis. One study demonstrated that oral valganciclovir is not inferior to intravenous ganciclovir for the treatment of CMV disease. [88]
Preemptive interventions
Using routine viral load monitoring and preemptive therapy, several studies have shown a reduction in PTLD prevalence based on historical controls, particularly in the pediatric liver transplant population. [11, 55, 82] These results have led many centers to perform routine viral load monitoring, particularly for preemptive interventions. These interventions have included reduction in immunosuppression or initiation of antiviral therapy when a significant rise in EBV viral load occurs. To date, no prospective clinical trials have been performed to confirm these results, but these types of studies are difficult to perform. The hypothesis that early detection leads to improved outcome remains also remains to be confirmed. Wagner et al have suggested that prompt intervention rather than preemptive intervention may be a better strategy in combination with viral load monitoring. [89]
Viral load monitoring provides evidence of EBV-infected B-cell proliferation but does not provide information about the host T-cell response. Studies of EBV-specific T-cell counts or cytotoxic T-cell assays may add further information to EBV viral loads. [90] . The serum levels of the B-cell homing chemokine CXCL13 have been shown to be elevated to greater degree in frank PTLD versus EBV reactivation. The serum CXCL13 levels also correlated with response to treatment and may thus serve as a useful surrogate marker if validated in further studies. [91]
EBV-specific inhibitors, including inhibitors of EBNA1 and EBNA2 as well as heat-shock protein 90 (HSP90) inhibitors have shown promise and are undergoing clinical trials. [92]
-
Epstein-Barr viral RNA can be detected by Epstein-Barr early region (EBER) assay (dark-blue staining cells) in the abnormal B cells from tumor masses of posttransplant lymphoproliferative disease (PTLD).








