Background
Ebstein anomaly is a rare congenital cardiac defect/malformation. The primary pathologic finding is abnormal development of the tricuspid valve marked by a downward displacement of the annular attachments of the septal and posterior leaflets of the tricuspid valve into the inlet portion of the right ventricle (see the image below). This downward displacement of the leaflets reduces the distal chamber of the right ventricle, leaving part of the ventricle above the valve as an extension of the right atrium. The entire wall of the right ventricle, both above and below the tricuspid valve, is often thin, dilated, and dysfunctional. In most patients, annular dilatation and malformation of the leaflets result in moderate-to-severe insufficiency of the tricuspid valve. Most patients have an atrial septal defect or a patent foramen ovale, which allows predominant right-to-left shunting at the atrial level. A high incidence of atrial and ventricular arrhythmia, including an association with Wolff-Parkinson-White Syndrome, occurs in these patients.
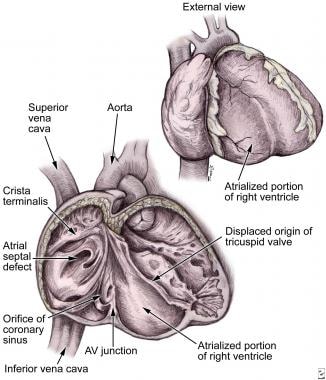 Anatomic features of Ebstein anomaly. Note the atrialized portion of the right ventricle and displacement of the tricuspid valve. AV = atrioventricular.
Anatomic features of Ebstein anomaly. Note the atrialized portion of the right ventricle and displacement of the tricuspid valve. AV = atrioventricular.
Ebstein anomaly is characterized by remarkable morphologic variability and a broad spectrum of clinical presentations. Consequently, the diagnosis of Ebstein anomaly may be made in symptomatic newborn infants, young adults, or middle-aged adults, depending on the severity of the defect and clinical manifestations.
This defect accounts for less than 1% of all congenital heart disease. [1] Geographic variation in the prevalence of this defect, inside or outside the United States, has not been documented. Although no increase in prevalence has been documented, an improvement in diagnostic techniques has led to earlier diagnosis. The anomaly occurs in both sexes with equal frequency.
History of the surgical procedure
In 1866, Wilhelm Ebstein, a young physician in Breslau, Poland, reported findings from a postmortem examination performed 2 years earlier. [2] The patient was a 19-year-old laborer who presented with dyspnea and palpitations and was noted to be profoundly cyanotic. In his report, Ebstein described in great detail the morphology and clinical correlations of the congenital cardiac malformation that bears his name. His report provided a strong basis for the subsequent development of surgical techniques for the treatment of this anomaly 100 years later. [3]
Early surgical attempts to treat Ebstein anomaly using palliative shunts resulted in extremely high mortality rates. [4] In 1958, Hunter and Lillihei described a technique of surgical repair that involved plication of the atrialized right ventricle, closure of the atrial septal defect, and tricuspid valve annuloplasty. [5] In 1964, Hardy reported the first successful repair of Ebstein anomaly using this technique. [6]
During the 1960s, most attempts to repair the tricuspid valve were unsuccessful, and prosthetic valve replacement became the preferred approach. In 1962, Christian Barnard described the first successful tricuspid valve replacement in a patient with Ebstein anomaly using a mechanical valve. [7] In the early 1970s, interest once again focused on tricuspid valve repair with a successful technique described by Danielson and colleagues at the Mayo Clinic. [8] Several modifications of tricuspid valve repair have been described recently, and early results have been successful. [9, 10, 11, 12]
Relevant Anatomy
The characteristic finding of Ebstein anomaly is a downward displacement of the attachments of the septal and posterior leaflets of the tricuspid valve below the true tricuspid annulus. These valve leaflets are often hypoplastic and adherent to the endocardial surface of the right ventricle. The commissure between these 2 leaflets is usually the point of maximal displacement. The anterior leaflet is typically larger than normal and sail-like in appearance. The anterior leaflet may be thin and fenestrated, and it may also adhere to the endocardial surface of the right ventricle. Rarely, a leaflet can divide, resulting in a double-orifice valve. [13]
The individual valve leaflets may be larger or smaller than expected. The chordae tendineae and papillary muscles are often abnormal in their number, development, and location. In rare instances, the anterior leaflet may attach to the apical region of the right ventricle and obstruct blood flow through the right ventricular outflow tract. Additionally, the tricuspid annulus is usually enlarged to 2-4 times the normal size.
Anatomic features of Ebstein anomaly are presented in the image below.
Anatomic features of Ebstein anomaly. Note the atrialized portion of the right ventricle and displacement of the tricuspid valve. AV = atrioventricular.
The displacement of the septal and posterior leaflets of the tricuspid valve into the right ventricle leaves a portion of the right ventricular wall between the leaflet attachments and the true annulus in continuity with the right atria. This atrialized right ventricle is usually very thin and dilated with a decrease in the actual number of myocardial fibers. The functional right ventricle below the valve may also be thin and dilated with decreased contractile function.
The anatomy of the conduction system is normal; the atrioventricular node is located at the apex of the triangle of Koch, and the sinoatrial node is located at the junction of the superior vena cava and the right atrium. The location of the bundle of His is normal, but abnormalities of the right bundle branch have been described. Accessory atrioventricular connections are present in as many as 20% of patients and are associated with Wolff-Parkinson-White syndrome. [14, 15]
The right atrium is enlarged and may reach enormous size. An atrial septal defect is usually present and ranges from a patent foramen ovale to a large secundum defect. The coronary arteries are normal in distribution, and the right coronary artery is often displaced by the enlarged right atrium and atrialized right ventricle. Associated cardiac anomalies include pulmonary stenosis or atresia, ventricular septal defect, patent ductus arteriosus, tetralogy of Fallot, and coarctation of the aorta. [4, 16] In patients with Wolff-Parkinson-White syndrome, accessory pathways are typically the atrioventricular type and are localized to the right ventricular free wall or to the posterior interventricular septum (bundles of Kent).
Ebstein anomaly of the left atrioventricular valve occurs in up to 75% of patients with L-transposition of the great arteries or corrected transposition of the great arteries. In these patients, displacement of the septal and posterior leaflets is similar to that in right-sided lesions, but the anterior leaflet is usually smaller. [17] In addition, the wall of the functional right ventricle is rarely thin and dilated, and the wall of the atrialized right ventricle is also less thin and dilated. Left-sided accessory pathways have been described only in those patients with Ebstein anomaly of the left-sided atrioventricular valve.
Pathophysiology
In most patients, the tricuspid valve is incompetent with some degree of functional impairment of the right ventricle. The atrialized right ventricle paradoxically moves with right atrial and right ventricular contractions. The net effect is reduced forward blood flow through the right ventricle and pulmonary arteries. The impaired filling of the functional right ventricle and the incompetence of the tricuspid valve both result in systemic venous hypertension.
The right atrium and the atrialized right ventricle become dilated, often to extreme proportions. In most patients, right-to-left shunting occurs across a defect in the atrial septum and results in cyanosis. The presence of pulmonary hypertension in neonates increases this atrial shunting and can lead to profound cyanosis in newborn infants. In some neonates, right ventricular outflow tract obstruction and pulmonary stenosis or atresia may result in a completely ductal-dependent pulmonary circulation. Both atrial arrhythmias and ventricular arrhythmias may contribute to impaired right ventricular function.
Although the primary pathology involves the right ventricle, patients with Ebstein anomaly may also demonstrate abnormal left ventricular geometry and function. The severity of left ventricular dysfunction is associated with the degree of displacement of the tricuspid valve, the size and dysfunction of the right ventricle, and the severity of paradoxical motion of the interventricular septum.
Etiology
The exact embryologic cause of Ebstein anomaly is unknown. The tricuspid valve leaflets form by a process of delamination of the inner layers of the inlet portion of the right ventricle. Evidence suggests that the anterior leaflet forms earlier in development than the septal and posterior leaflets. In Ebstein anomaly, the insertions of the septal and posterior leaflets are displaced to the junction of the inlet and trabecular portions of the right ventricle, indicating abnormal delamination. [17] The insertion of the anterior leaflet is at the level of the true annulus. The right ventricle endocardium is often thickened and fibrotic, suggesting that formation of the valve leaflets was interrupted prior to completion of the delamination process. The tricuspid valve is usually incompetent, but it may also be stenotic or even imperforate. In addition, both the atrialized right ventricular wall and the functional right ventricular wall may be abnormally thin and fibrotic.
The observed morphologic variability indicates the complexity of this defect's origin during embryologic development. Interestingly, exposure of the fetus to lithium carbonate during the first trimester has been linked to the development of Ebstein anomaly. [18] Although no specific genetic inheritance pattern has been documented, a familial association has been reported. [4]
Prognosis
Early surgical experience with Ebstein anomaly was associated with high mortality rates in excess of 20%. Over the years, mortality has improved as the disorder has become well understood. Studies from the Mayo Clinic indicate that risk factors for early mortality include body weight less than 10.7 kg (23.6 lb) and age younger 2.5 years. [19]
Over the past 2 decades, the actuarial survial at 5 and 10 years is 92% and 90%, respectively. Another study involving 113 surgical patients revealed a much lower mortality and a higher 5-year survival in children undergoing a bidirectional Glenn compared to children who did not undergo the shunt. [20]
Patients who have moderate to severe tricuspid regurgitation at discharge are at a high risk for reoperation. [21]
Reports indicate that radiofrequency ablation of Wolff-Parkinson-White syndrome in patients with Ebstein anomaly is associated with a lower success rate compared to patients who do not have the congenital heart disorder. [22]
Complications
Potential complications of Ebstein anomaly include the following:
-
Arrhythmias
-
Sudden death
-
Heart failure
-
Bacterial endocarditis
-
Paradoxical emboli
-
Stroke
Patient Education
Parents and/or patients with Ebstein anomaly should be informed that surgery is often required in the presence of symptoms. However, some infants with severe heart dysfunction may not be candidates for surgical repair but may be better suited for a heart transplant. In addition, for those patients who do undergo repair of the anomaly, multiple surgeries may be required in the future.
For patient education resources, see the Heart Health Center, as well as Tetralogy of Fallot, Palpitations (Causes, During Pregnancy, Symptoms, Treatment, Ventricular Septal Defect, Atrial Fibrillation (AFib), Congestive Heart Failure, Pleurisy (Pain, Symptoms, Causes, Treatment, and High Blood Pressure (Symptoms, Sings, Causes, Diet, Medication).
-
Anatomic features of Ebstein anomaly. Note the atrialized portion of the right ventricle and displacement of the tricuspid valve. AV = atrioventricular.
-
Characteristic chest radiograph of a neonate with Ebstein anomaly. The heart shadow demonstrates cardiomegaly, with evidence of severe right atrial enlargement.
-
Surgical repair of Ebstein anomaly as described by Danielson. (A) The right atrium is incised and reduced, and the atrial septal defect is closed with a patch. The arrow identifies the large anterior leaflet. (B) Mattress sutures with felt pledgets are used to pull the tricuspid annulus and valve together in a horizontal plane, obliterating the atrialized right ventricle. (C) Sutures are tied after all have been inserted. The arrow identifies the septal leaflet. (D) A posterior annuloplasty is used to narrow the orifice of the tricuspid annulus. (E) Completed repair, resulting in a competent tricuspid valve.
-
Surgical repair of Ebstein anomaly as described by Carpentier. The anterior and posterior leaflets are detached from the tricuspid annulus. In type D lesions, fenestrations are used to create interchordal spaces for the passage of blood into the right ventricle outflow tract (insert). Mattress sutures with pledgets are placed in a vertical plane to plicate the atrialized portion of the right ventricle (top right). The anterior leaflet is reattached at the level of the true annulus with a continuous running suture (bottom left). An annuloplasty ring is inserted to reinforce the repair (bottom right).
-
Surgical repair of Ebstein anomaly in the neonate as described by Starnes. The atrial septal defect is enlarged by excising the remaining septum. The tricuspid valve orifice is closed with a Gore-Tex patch, effectively creating tricuspid atresia. A Gore-Tex shunt (not shown) is then placed to connect the innominate artery to the right pulmonary artery. PTFE = polytetrafluoroethylene.
-
Surgical replacement of the tricuspid valve in Ebstein anomaly. (A) The atrialized right ventricle is plicated in a horizontal plane. (B) Sutures are placed on the atrial side of the coronary sinus and atrioventricular node to avoid injury to the conduction system. (C) Sutures are tied with the heart beating and perfused to ensure the conduction system is intact.
-
Surgical replacement of the tricuspid valve using a pericardial patch to avoid injury to the conduction system. The valve insertion is begun anterior to the coronary sinus using a continuous running suture. A glutaraldehyde-treated pericardial patch is sutured to the septal portion of the prosthetic valve sewing annulus. The free margin of the patch is then sutured to the atrial tissue beyond the area of the conduction tissue. AV = atrioventricular.
-
Operative steps for Ebstein anomaly repair. (A) Opened right atrium showing displacement of the tricuspid valve. ASD = atrial septal defect, CS = coronary sinus, TTA = true tricuspid annulus. (B) Detached part of the anterior and posterior leaflet as a single piece. (C) Clockwise rotation of the posterior leaflet edge to be sutured to the anterior leaflet septal edge and plication of the true tricuspid annulus to bring the valve to a uniform level. (D) Completion of valve attachment to the true tricuspid annulus and closure of the atrial septal defect. Used with permission from Elsevier (Fig 1 from Da Silva JP, Baumgratz JF, da Fonseca L, et al. The cone reconstruction of the tricuspid valve in Ebstein's anomaly. The operation: early and midterm results. J Thorac Cadiovasc Surg. 2007 Jan;133(1):215-23).
-
The cardiac silhouette is usually enlarged in Ebstein anomaly. A chest radiograph will usually show an enormous shadow of the right atrium.
-
Pathology of Ebstein anomaly: Dilated right atrium (RA) with inferior displacement of an abnormal tricuspid valve (TV) septal leaflet, which results in a small right ventricle (RV) that is atrialized. ASD = atrial septal defect, LA = left atrium, LV = left ventricle, PA = pulmonary artery, R-L = right to left.





