Background
Relatively speaking, congenital duodenal atresia is one of the more common intestinal anomalies treated by pediatric surgeons, occurring 1 in 5,000-10,000 live births. [1] In 25-40% of cases, the anomaly is encountered in an infant with trisomy 21 (Down syndrome). [2] The definitive intervention to correct the anomaly in the newborn is surgical and typically consists of duodenoduodenostomy.
History of the Procedure
Calder published the first report of duodenal obstruction in 1733 when he described two children with "preternatural confirmation of the guts." [3] Both infants died, as did infants with this defect subsequently reported. Scattered reports of duodenal obstruction appeared in the European literature over ensuing years. In 1916, the first survivor was reported, yet survival in the early 20th century remained rare.
Significant improvement in morbidity and mortality has occurred only over the last 50 years. [4] Because of progress in pediatric anesthesia, neonatology, and surgical techniques, current survival is about 90% in infants who present with duodenal obstruction. At present, the standard operative procedure consists of duodenoduodenostomy via a right supraumbilical incision, although more recent advancements have enabled surgeons to repair the defect by minimally invasive means. [5, 6, 7]
Relevant Anatomy
Duodenal atresia is subcategorized into three types [1, 8] :
-
Type 1 (most common): A mucosal web or diaphragm obstructs the duodenal lumen, causing proximal dilatation and elongation of the web. Due to the stretching and elongation of the diaphragm, the mucosal web occasionally has the appearance of a windsock. The true site of obstruction will be proximal to the distal aspect of the dilated bowel. The web may also be fenestrated, leading to only a partial obstruction.
-
Type 2: The proximal and distal segments of the duodenum are connected by an atretic cord.
See the following image.
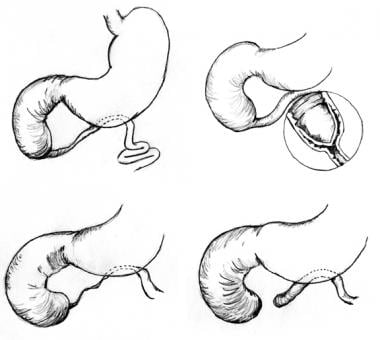 Pediatric duodenal atresia. Three anatomic types of duodenal atresia are recognized. In type 1 atresia, a membrane traverses the internal diameter of the duodenum. This membrane may be elongated, giving rise to the windsock type 1 duodenal atresia. In type 2 atresia, the atretic ends of the duodenum are connected by a fibrous cord. In type 3 atresia, the atretic segments are completely separated.
Pediatric duodenal atresia. Three anatomic types of duodenal atresia are recognized. In type 1 atresia, a membrane traverses the internal diameter of the duodenum. This membrane may be elongated, giving rise to the windsock type 1 duodenal atresia. In type 2 atresia, the atretic ends of the duodenum are connected by a fibrous cord. In type 3 atresia, the atretic segments are completely separated.
Problem
The differential diagnosis of neonatal upper gastrointestinal obstruction or vomiting includes the following:
-
Duodenal atresia and stenosis
-
Antral web
-
Annular pancreas
-
Preduodenal portal vein
-
Any intestinal atresia
-
Duodenal duplication
Duodenal obstructions can be complete or partial. Intrinsic duodenal obstructions may be attributed to atresia or mucosal webs. Even with annular pancreas, the obstruction is due to an associated intrinsic narrowing of the duodenum within the annular portion. Duodenal extrinsic obstruction can occur in association with malrotation or a preduodenal portal vein.
Duodenal atresia can take many forms, but proximal and distal intestinal segments always end blindly. [11] The intestine on either side of the defect may be in apposition (type 1), or separated by a fibrous cord (type 2), or a gap (type 3). [12] Hallmarks of duodenal atresia are a dilated proximal segment and a decompressed distal segment. Although obstruction may occur anywhere within the duodenum, it is most common in the vicinity of the ampulla of Vater.
Stenosis may manifest as a stricture or a fenestrated intraluminal diaphragm (ie, web). The fenestration within the diaphragm is usually singular and centrally located within the lumen of the duodenum, although variations have been reported. A windsock abnormality is a thin diaphragm that has ballooned distally as a result of peristalsis. Together, both duodenal atresia and stenosis are a common etiology of intestinal obstruction in the newborn. [5]
Pathophysiology
Duodenal maldevelopment occurs secondary to either inadequate endodermal proliferation (gut elongation outpaces proliferation) or failure of the epithelial solid cord to recanalize (failure of vacuolization). Multiple investigators have demonstrated that the epithelium of the duodenum proliferates during days 30-60 of gestation, completely plugging the duodenal lumen. A subsequent process termed vacuolization occurs whereby the solid duodenum is recanalized. Vacuolization is believed to occur by way of apoptosis, or programmed cell death, which occurs during normal development within the lumen of the duodenum. Occasionally, duodenal atresia is associated with annular pancreas—pancreatic tissue that surrounds the entire circumference of the duodenum. This is likely due to failure of duodenal development rather than robust and/or abnormal growth of the pancreatic buds and can also be associated with other biliary anomalies. [9, 13]
At the cellular level, the gastrointestinal (GI) tract develops from the embryonic gut, which is composed of an epithelium derived from endoderm, surrounded by cells of mesodermal origin. Cell signaling between these two embryonic layers appears to play a critical role in coordinating patterning and organogenesis of the duodenum. Sonic hedgehog genes encode members of the hedgehog family of cell signals. Both are expressed in gut endoderm, whereas target genes are expressed in discrete layers in the mesoderm. Mice with genetically altered sonic hedgehog signaling display duodenal stenosis, which suggests that genetic defects in the sonic hedgehog family of genes may influence the development of duodenal abnormalities.
Epidemiology
Reported incidence rates for duodenal atresia range from 1 in 5,000 to 10,000 live births; rates published in the United States and internationally do not appear to differ. [14] Duodenal atresia is not usually regarded as a familial condition, despite isolated reports of this condition in multiple siblings.
Etiology
Although the underlying cause of duodenal atresia remains unknown, its pathophysiology has been well described. Frequent association of duodenal atresia or stenosis with other neonatal malformations suggests both anomalies are due to a development error in the early period of gestation. However, duodenal atresia differs from other atresias of the small and large bowel, which are isolated anomalies caused by mesenteric vascular accidents during later stages of development. No predisposing maternal risk factors are known. Although 25-40% of patients with duodenal atresia have Down syndrome (trisomy 21), this condition is not an independent risk factor for developing duodenal atresia. [2, 5, 15]
Presentation
Duodenal atresia is a disease of newborn infants. Cases of duodenal stenosis or perforated duodenal web rarely remain undiagnosed until childhood or adulthood; these cases represent the exception rather than the rule. [16] Duodenal atresia appears to be equally distributed between infants of both sexes, with no reported predilection for one race.
The use of modern ultrasonography has allowed many infants with duodenal obstruction to be identified prenatally. [17] In a large cohort study of 18 different congenital malformation registries from 11 European countries, 52% of infants with duodenal obstruction were identified in utero. [18]
Duodenal obstruction is characterized by a double-bubble sign on prenatal ultrasonography. The first bubble corresponds to the stomach and the second to the post-pyloric dilated duodenum. Prenatal diagnosis allows the mother the opportunity to receive prenatal counseling and to consider delivery at or near a tertiary care facility that is able to care for infants with gastrointestinal (GI) anomalies. [18]
Presenting symptoms and signs of duodenal atresia are the result of proximal intestinal obstruction. Duodenal atresia is typically characterized by the onset of vomiting within hours of birth. Whereas the vomitus is most often bilious, it may be nonbilious because 15% of defects occur proximal to the ampulla of Vater. [19] Occasionally, infants with duodenal stenosis escape detection of an abnormality until childhood or, rarely, adulthood before a partial obstruction is noted. Nevertheless, one should assume any child with bilious vomiting has a proximal GI obstruction until proven otherwise, and further workup should be initiated expeditiously.
Once delivered, an infant with duodenal atresia typically has a scaphoid abdomen. One may occasionally note epigastric fullness from dilation of the stomach and proximal duodenum. Gastric aspiration volume in a newborn of more than 20 mL is suggestive of intestinal obstruction; normally, aspirates should be minimal. [20] There is usually no alteration of passing meconium within the first 24 hours of life. Dehydration, weight loss, and electrolyte imbalance soon follow unless fluid and electrolyte losses are adequately replaced. If intravenous hydration is not begun, a hypokalemic/hypochloremic metabolic alkalosis develops, as with other etiologies of proximal GI obstruction.
Timing of Surgical Repair
Although duodenal atresia is a surgically treated disease, operating on an infant with duodenal obstruction in the middle of the night is usually unnecessary. The exception is the infant with duodenal obstruction due to malrotation/volvulus, which can evaluated with an upper gastrointestinal contrast study.
Over 50% of infants with duodenal atresias have concomitant anomalies. The most common limitations that apply to timing the repair include stabilization of the fluid and electrolyte balance and exclusion of overwhelming congenital defects that would preclude the use of a general anesthetic (ie, complex congenital heart disease). Surgical correction can begin any time after these issues are addressed and optimized.
Prior to repair, infants can be maintained on orogastric suction and intravenous nutrition with aggressive repletion of fluid and electrolyte losses while any life-threatening issues are addressed.
Prognosis
The overall mortality for infants with duodenal atresia was 33% in a large series published in 1967. Today, the early mortality associated with this condition has declined to approximately 3% in most series.
Most deaths occurring in association with duodenal atresia are attributed to the presence of other associated anomalies (usually complex cardiac defects). Improvement in survival is most likely a result of advances in neonatal intensive care, nutritional support, and pediatric anesthesia. Long-term survival is excellent, at reported rates of greater than 90%.
-
Pediatric duodenal atresia. Incision for duodenal exposure.
-
Pediatric duodenal atresia. Side-to-side duodenoduodenostomy.
-
Pediatric duodenal atresia. Diamond-shaped duodenoduodenostomy.
-
Pediatric duodenal atresia. Three anatomic types of duodenal atresia are recognized. In type 1 atresia, a membrane traverses the internal diameter of the duodenum. This membrane may be elongated, giving rise to the windsock type 1 duodenal atresia. In type 2 atresia, the atretic ends of the duodenum are connected by a fibrous cord. In type 3 atresia, the atretic segments are completely separated.
-
Pediatric duodenal atresia. Upper gastrointestinal series demonstrating duodenal atresia.




