Background
Argininosuccinate (ASA) lyase deficiency results in defective cleavage of ASA. This leads to an accumulation of ASA in cells and an excessive excretion of ASA in urine. In virtually all respects, this disorder shares the characteristics of other urea cycle defects. [1] The most important characteristic of ASA lyase deficiency is its propensity to cause hyperammonemia in affected individuals. See the image below.
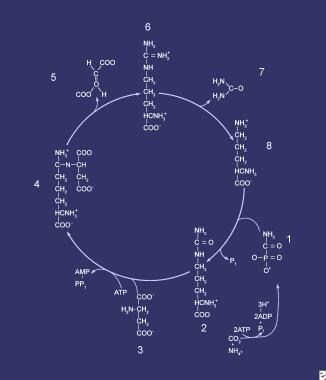 Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.
Pathophysiology
The hepatic urea cycle is the major route for waste nitrogen disposal; nitrogen generation results chiefly from protein and amino acid metabolism. Low-level synthesis of certain cycle intermediates in extrahepatic tissues makes a small contribution to waste nitrogen disposal. A portion of the cycle is mitochondrial in nature; mitochondrial dysfunction may impair urea production and may result in hyperammonemia. Overall, the cycle’s activity is regulated by the rate of synthesis of N -acetylglutamate, the enzyme activator that initiates incorporation of ammonia into the cycle.
The rate-limiting step is carbamoyl phosphate synthetase (CPS) disposal of waste nitrogen. However, in patients with a genetic deficiency in an additional enzyme in the cycle (other than CPS), the deficient enzyme becomes rate limiting. This occurs in patients with argininosuccinic aciduria, despite the fact that formation of this substance ensures incorporation of the 2 waste nitrogen molecules normally found in urea. Although failure to release the arginine limits the cycle rate and slows hepatic regeneration of the distal intermediates of the cycle, this is unlikely to entirely explain the clinical findings, because ASA is excreted by the kidney at a rate practically equivalent to the glomerular filtration rate (GFR). [2, 3, 4]
A potential alternative is that the inability to release arginine from ASA leads to arginine deficiency, in turn restricting both protein and nitric oxide synthesis, the latter exposing tissues to damage from oxide radical damage.
Whether ASA itself causes a degree of toxicity due to hepatocellular accumulation is unknown; such an effect could help explain hyperammonemia development in affected individuals. Regardless, the name of the disease is derived from the rapid clearance of ASA in urine, although elevated levels of ASA can be found in plasma. Hyperammonemia in this disease manifests with the typical findings and carries all of the attendant consequences associated with other urea cycle diseases.
Epidemiology
Frequency
United States
ASA lyase deficiency is rare,, affecting an estimated 1 in 70,000 live births. It is the second-most common urea cycle disorder. [5] As with other disorders in this category, this deficiency can manifest in neonates or later in life, and true incidence cannot be cited without population screening data. [6]
International
The Druze community in Israel has a carrier frequency of 1:41. [7] According a study of urea cycle diseases in Finland, 20 cases of ASA lyase deficiency had been reported by 2007. [8]
Mortality/Morbidity
ASA lyase deficiency is associated with high mortality and morbidity rates. Failure to suspect hyperammonemia and to obtain blood ammonia levels results in certain morbidity and, likely, death because routine laboratory test findings are unrevealing.
Sex
Inherited as an autosomal recessive trait, argininosuccinic aciduria affects both sexes equally.
Prognosis
Prognosis is guarded.
Although intellectual impairment is the rule, even among patients who receive excellent and timely treatment, some patients with ASA lyase deficiency reportedly develop normally.
Patient Education
Advise parents of an affected infant that they are obligate heterozygotes because the disease is inherited as an autosomal recessive trait. This trait leads to a recurrence risk of 1:4 (25%) with each subsequent pregnancy.
Prenatal diagnosis is available for ASA lyase deficiency, although the involved diagnostic procedures are not trivial. Even in cases in which elective abortion is not an option, parents should be prepared for an affected infant in order to avoid early hyperammonemia.
Advise parents to scrupulously follow the dietary and medication instructions and to seek early medical attention for all intercurrent illnesses.
-
Compounds comprising the urea cycle are numbered sequentially, beginning with carbamyl phosphate (1). At this step, the first waste nitrogen is incorporated into the cycle; at this step, N-acetylglutamate exerts its regulatory control on the mediating enzyme, carbamyl phosphate synthetase (CPS). Compound 2 is citrulline, the product of condensation between carbamyl phosphate (1) and ornithine (8); the mediating enzyme is ornithine transcarbamylase. Compound 3 is aspartic acid, which is combined with citrulline to form argininosuccinic acid (ASA) (4); the reaction is mediated by ASA synthetase. Compound 5 is fumaric acid generated in the reaction that converts ASA to arginine (6), which is mediated by ASA lyase.



