Background
Tricuspid atresia may be defined as congenital absence or agenesis of the tricuspid valve. [1] It is the third most common cyanotic congenital heart defect; the other 2 frequently observed cyanotic congenital cardiac anomalies are transposition of the great arteries and tetralogy of Fallot. Tricuspid atresia is the most common cause of cyanosis with left ventricular hypertrophy. [2]
Although some authors state that Holmes (1824) or Kuhne (1906) first described tricuspid atresia, [3] Rashkind's methodical and thorough historical review indicates that Kreysig (1817) reported the first case in 1817. [4] An 1812 report by the editors of the London Medical Review (1812) appears to fit the description of tricuspid atresia, but they did not use this specific term. [4]
Terminology
Little more than 3 decades ago, the terminology for this defect (eg, tricuspid atresia, univentricular heart, univentricular atrioventricular connection) was intensely debated. [5, 6, 7, 8, 9, 10, 11] This debate was summarized in a 1990 issue of The American Journal of Cardiology, [12] in which Rao offered strong evidence and argued on the basis of data that Bharati and Lev, [10, 11] Wenink and Ottenkamp, [13] Gessner, [14] and Rao gathered support for tricuspid atresia as the correct and logical term to describe this well-characterized pathologic and clinical condition.
Embryology
The atrioventricular valves develop shortly after the atrioventricular canal divides. The tricuspid valve leaflets have several origins. The septal leaflet of the tricuspid valve mostly develops from the inferior endocardial cushion with a small contribution from the superior cushion. The anterior and posterior tricuspid valve leaflets develop by undermining of a skirt of ventricular muscle tissue. The process of undermining extends until the atrioventricular valve junction is reached. Resorption of the muscle tissue produces normal-appearing valve leaflets and chordae tendineae. [15, 16, 17] Fusion of developing valve leaflet components results in stenosis (partial fusion) or atresia (complete fusion) of the valve. [17, 18]
Whether a muscular type of tricuspid atresia develops or whether well-formed but fused tricuspid-valve leaflets develop depends on the stage of development when the embryologic aberration takes place. [17, 18] The classic muscular form of tricuspid atresia develops if the embryologic insult occurs early in gestation, and fused valve leaflets occur if the embryologic abnormality occurs slightly later than this in gestation. If the valve fusion is incomplete, stenosis of the tricuspid valve develops.
The pathologic, clinical, and electrocardiographic features of tricuspid stenosis and atresia are similar. [19] Therefore, the fact that isolated congenital tricuspid stenosis belongs to the group of tricuspid atresia defects and that their embryologic developments are similar is no surprise. Thus, the tricuspid valve stenosis, tricuspid atresia with well-formed but fused valve leaflets, and the muscular type of tricuspid atresia represent a spectrum of morphologic abnormalities. [12, 18]
Anatomy
The pathologic anatomy of tricuspid atresia is best understood by reviewing variations in valvar morphology.
The most common type of tricuspid atresia is muscular (see the image below). [9, 20] It is characterized by a dimple or a localized fibrous thickening in the floor of the right atrium at the expected site of the tricuspid valve. The muscular variety accounts for 89% of cases. [6]
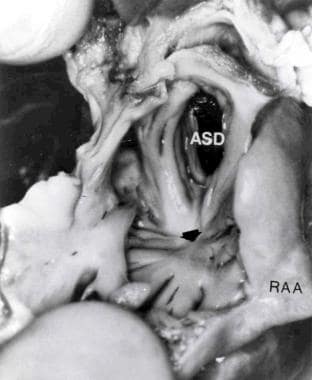 Cardiac specimen from a patient with the muscular type of tricuspid atresia. The right atrium was opened by cutting through the right atrial appendage (RAA). Note the dimple (arrow) in the floor of the right atrium with muscle fibers radiating around it. An atrial septal defect (ASD) is also shown. From Rao PS, Levy JM, Nikicicz E, Gilbert-Barness EF. Tricuspid atresia: association with persistent truncus arteriosus. Am Heart J 1991, 122:829, with permission.
Cardiac specimen from a patient with the muscular type of tricuspid atresia. The right atrium was opened by cutting through the right atrial appendage (RAA). Note the dimple (arrow) in the floor of the right atrium with muscle fibers radiating around it. An atrial septal defect (ASD) is also shown. From Rao PS, Levy JM, Nikicicz E, Gilbert-Barness EF. Tricuspid atresia: association with persistent truncus arteriosus. Am Heart J 1991, 122:829, with permission.
In the membranous type (6.6%), the atrioventricular portion of the membranous septum forms the floor of the right atrium at the expected location of the tricuspid valve. This particular type appears to be associated with absent pulmonary valve leaflets.
Minute valvar cusps are fused together in the valvar type (1%).
In the Ebstein type (2.6%), fusion of the tricuspid valve leaflets occurs; attachment is displaced downward, and plastering of the leaflets to the right ventricular wall occurs. [21] This variant is rare but well documented.
The atrioventricular canal type is extremely rare (0.2%). In this type, a leaflet of the common atrioventricular valve seals off the only entrance into the right ventricle. [13]
In the final type, unguarded with a muscular shelf (0.6%), the atrioventricular junction is unguarded, but the inlet component of the morphologic right ventricle is separated from its outlet by a muscular shelf. [22]
The right atrium is enlarged and hypertrophied. An interatrial communication is necessary for survival. This communication most commonly is a stretched patent foramen ovale. Sometimes, an ostium secundum or an ostium primum atrial septal defect (ASD) is present. In rare cases, the patent foramen ovale is obstructive and may form an aneurysm of the fossa ovalis, which is sometimes large enough to produce mitral inflow obstruction. The left atrium may be enlarged, especially when the pulmonary blood flow is increased. The mitral valve is morphologically normal; it is rarely incompetent and has a large orifice. The left ventricle is enlarged and hypertrophied but often morphologically normal.
The ventricular septal defect (VSD) is usually small; however, it can be large, or several VSDs may be present. [23] The ventricular septum is rarely intact. When present, the VSD may be conoventricular or perimembranous in type (inferior to the septal band), it may be of conal septal malalignment type (between the limbs of the septal band), or it may be of the muscular or atrioventricular canal type. [24, 25] Muscular VSDs are the most common defects and are usually restrictive; they produce subpulmonary stenosis in patients with normally related great arteries and simulate subaortic obstruction in patients with transposition of the great arteries. [26, 27]
The right ventricle is small and hypoplastic, and its size largely depends on the anatomic type. [28] In patients with a large VSD or transposition of the great arteries, the size of the right ventricle may be larger, but, even in these patients, the right ventricle is smaller than normal. In patients with pulmonary atresia and normally related great arteries, the right ventricle is small and may escape detection. However, it is a true right ventricle in most patients; it is composed of a sharply demarcated infundibulum with septal and parietal bands and a sinus with trabeculae, which may communicate with the left ventricle by means of a VSD. By definition, the inflow region is absent, although papillary muscles may occasionally be present.
The great artery relationship is variable and forms the basis of a major classification and will be described in the next section. Obstruction to the pulmonary outflow tract is present in most cases of tricuspid atresia and is used in the scheme of classification. The aorta is either normal or slightly larger than normal. In 30% of patients, various associated cardiac defects are present; aortic coarctation and persistent left superior vena cava are particularly notable.
Associated cardiac defects in tricuspid atresia outlined below. [29]
Defects that form the basis for classification are as follows:
-
D-Transposition of the great arteries
-
L-Transposition of the great arteries
-
Double outlet left ventricle
-
Other malpositions of the great arteries
-
Truncus arteriosus [30]
Defects that may need attention before or during palliative or total surgical correction are as follows:
-
Absent pulmonary valve
-
Aneurysm of the atrial septum
-
Anomalous origin of the coronary arteries from the pulmonary artery
-
Anomalous origin of the left subclavian artery
-
Anomalous origin of the right subclavian artery
-
Aortopulmonary fistula
-
Coarctation of the aorta
-
Common atrium
-
Cor triatriatum dexter
-
Coronary sinus atrial septal defect
-
Double aortic arch
-
Double-outlet left atrium
-
Hemitruncus
-
Hypoplastic ascending aorta and/or aortic atresia
-
Ostium primum ASD
-
Parchment right ventricle
-
Patent ductus arteriosus
-
Persistent left superior vena cava
-
Right aortic arch
-
Subaortic stenosis
-
Total anomalous pulmonary venous connection
-
Tubular hypoplasia of the aortic arch
-
Valvar aortic stenosis
Other associated defects are as follows:
-
Juxtaposition of the atrial appendages
-
Anomalous entry of coronary sinus into the left atrium
Classification
Tricuspid atresia is classified according to the morphology of the valve, [20, 31] the radiographic appearance of pulmonary vascular markings, [32, 33] and the associated cardiac defects. [3, 34, 35, 36, 37]
Van Praagh and associates (1971) initially proposed a classification based on the morphology of the atretic tricuspid valve. [20] He and others later modified and expanded the classification, as described in Tricuspid Atresia. [1, 6] All other morphologic types are described above in the Anatomy section. For pathologic, echocardiographic, and angiographic examples, particularly the rare anatomic types, the interested reader is referred to Tricuspid Atresia [6] and the Atlas of Heart Disease: Congenital Heart Disease. [1]
Astley and associates (1953) proposed a classification based on pulmonary vascular markings on a chest radiograph: Group A are cases with decreased pulmonary vascular markings, and group B are those with increased pulmonary vascular markings. [32] Dick et al (1975) added a third group, group C, to describe cases with a transition from increased to decreased pulmonary vascular markings. [33] This type of classification has some clinical value, although a more precise definition than these can often be made by using noninvasive 2-dimensional (2D) and Doppler echocardiography.
In 1906, Kuhne first proposed a classification based on great-artery relationships, [3] which Edwards and Burchell expanded in 1949. [34] Keith, Rowe, and Vlad popularized this classification in 1967. [35] Other investigators have offered various other classifications. These are reviewed in detail in the American Heart Journal [37] and Tricuspid Atresia. [7] Although these classifications are generally good, their exclusion of some variations in great-artery relationships and the lack of consistency in subgroups are problematic. Therefore, the following comprehensive-yet-unified classification was proposed [37] :
The principle grouping continues to be based on the following interrelationships of the great arteries:
-
Type I - Normally related great arteries
-
Type II - D-Transposition of the great arteries
-
Type III - Great artery positional abnormalities other than D-transposition of the great arteries: (1) Subtype 1 involves L-transposition of the great arteries, (2) subtype 2 involves double outlet right ventricle, (3) subtype 3 involves double outlet left ventricle, (4) subtype 4 involves D-malposition of the great arteries (anatomically corrected malposition), and (5) subtype 5 involves L-malposition of the great arteries (anatomically corrected malposition)
-
Type IV - Persistent truncus arteriosus
All types and subtypes are subdivided into the following subgroups:
-
Subgroup a - Pulmonary atresia
-
Subgroup b - Pulmonary stenosis or hypoplasia
-
Subgroup c - No pulmonary stenosis (normal pulmonary arteries)
After the above categorization, the status of the ventricular septum (intact or VSD) and the presence of other associated malformations are described.
This unified classification includes all the previously described abnormalities in the positions of the great arteries and can be further expanded if new variations are revealed. This classification maintains uniformity of the subgroups and preserves the basic principles of classification that Kuhne, Edwards and Burchell, and Keith, Rowe, and Vlad devised.
Pathophysiology
Prenatal circulation
Despite the clinically significant alterations in fetal circulation in tricuspid atresia, such changes are not detrimental to normal fetal development.
In a fetus with a normally developed heart, a substantial portion of the highly saturated blood in the inferior vena cava, which carries umbilical venous return from the placenta, is diverted into the left atrium through the patent foramen ovale. From there, it traverses into the left ventricle and aorta. Thus, the brain and heart receive blood with a high partial pressure of oxygen (PO2). [38, 39] In the normal fetus, the desaturated blood in the superior vena cava passes through the tricuspid valve, right ventricle, and pulmonary artery. Because of high pulmonary vascular resistance (PVR), the desaturated blood is then diverted through the ductus arteriosus into the descending aorta and umbilical arteries. The blood then returns to the placenta for oxygenation. [38, 39]
In tricuspid atresia, blood from both venae cavae is forced across the patent foramen ovale into the left heart. As a consequence, the PO2 differential present in a normally developed fetus is not present in the fetus with tricuspid atresia. The lowered PO2 to the brain and heart and elevated PO2 to the lungs do not seem to produce clinically discernible postnatal abnormalities. [18, 38, 39]
In patients with tricuspid atresia and associated pulmonary atresia (types Ia and IIa), the pulmonary blood flow is supplied entirely through the ductus arteriosus. Therefore, the ductus only carries 8-10% of combined ventricular output compared with 66% of combined ventricular output in a normally developed fetus. Also, acute angulation of the ductus arteriosus occurs at its origin because of reversed direction of ductal flow. These 2 factors may make the ductus arteriosus less responsive to postnatal stimuli than it usually is.
In a fetus with tricuspid atresia type I anatomy and a small or absent VSD (types Ia and Ib), almost all the left ventricular output is ejected into the aorta and transported down to the placenta. As a consequence, the isthmus of the aorta carries a larger-than-normal proportion of cardiac output; this is thought to be the reason for rarity of aortic coarctation in this subset of patients with tricuspid atresia.
In contrast, in patients with tricuspid atresia type II (transposition of the great arteries), an increased portion of the blood goes through the ductus arteriosus into the descending aorta. Therefore, the flow across the aortic isthmus is minimal, which accounts for the relatively high incidence of aortic coarctation in this subset of patients. [18, 38, 39]
Postnatal circulation
Because of the atretic tricuspid valve, all systemic venous blood must be shunted across the interatrial septal communication into the left atrium. This obligatory shunting causes admixture of all systemic venous and pulmonary venous returns. This blood then passes onto the left ventricle across the mitral valve. [18, 38, 39] This flow pattern occurs in all types but type III subtypes 1 and 5. In these exceptions, the atretic morphologic tricuspid valve is left sided because of ventricular inversion; therefore, the pathophysiology is that of mitral atresia with consequent left-to-right shunting of pulmonary venous return. [18, 39]
In patients with normally related great arteries (type I) and a VSD, shunting across the VSD permits perfusion of the lungs. In the absence of VSD, pulmonary blood flow is derived through patent ductus arteriosus or aortopulmonary collateral vessels. [18, 38, 39] Some means of lung perfusion is crucial for patient survival. The systemic blood flow is derived directly from the left ventricle.
In patients with D-transposition of the great arteries (type II), the lungs receive the blood flow from the left ventricle. The aorta receives blood from the left ventricle via the VSD and right ventricle. [18, 38, 39] In other types of tricuspid atresia, the routes of aortic and pulmonary artery flow depend on the size of the VSD and associated cardiac defects.
Other physiologic principles
Arterial desaturation
Systemic arterial desaturation is present in all patients with tricuspid atresia because of obligatory admixture of the systemic, coronary, and pulmonary venous returns in the left atrium. The degree of arterial desaturation depends on the amount of pulmonary blood-flow. [18, 39] The arterial oxygen saturation has a curvilinear relationship (see the image below), with a pulmonary-to-systemic blood flow ratio (Qp:Qs) that reflects the pulmonary blood flow. A Qp:Qs ratio of 1.5-2.5 seems to result in adequate oxygen saturation. Higher pulmonary flow does not significantly increase oxygen saturation but instead produces left ventricular volume overloading.
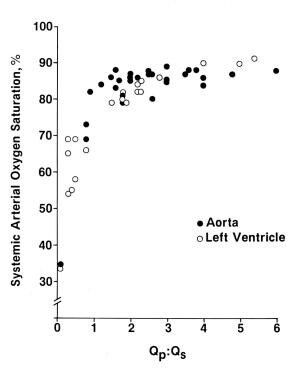 Systemic arterial saturations. Left ventricular (LV) and aortic (Ao) values are plotted against the pulmonary-to-systemic blood-flow ratio (Qp:Qs). Both type I and type II anatomy are included. Note the curvilinear relationship between the parameters. At low Qp:Qs levels, a slight increase in Qp:Qs produces large increase in systemic oxygen saturation; at high Qp:Qs levels, a further increase does not produce a notable increase in oxygen saturation. The ideal Qp:Qs appears to be 1.5-2.5, which results in oxygen saturations in the low 80s. From Rao PS. Cardiac catheterization in tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co: 1982:153, with permission.
Systemic arterial saturations. Left ventricular (LV) and aortic (Ao) values are plotted against the pulmonary-to-systemic blood-flow ratio (Qp:Qs). Both type I and type II anatomy are included. Note the curvilinear relationship between the parameters. At low Qp:Qs levels, a slight increase in Qp:Qs produces large increase in systemic oxygen saturation; at high Qp:Qs levels, a further increase does not produce a notable increase in oxygen saturation. The ideal Qp:Qs appears to be 1.5-2.5, which results in oxygen saturations in the low 80s. From Rao PS. Cardiac catheterization in tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co: 1982:153, with permission.
Pulmonary blood flow
The clinical features of tricuspid atresia largely depend on the quantity of pulmonary blood flow. [2, 18, 40, 41, 42] A neonate with markedly decreased pulmonary flow is likely to present early in the neonatal period with signs of severe cyanosis, hypoxemia, and acidosis. On the contrary, if the pulmonary blood flow is increased, the neonate may not appear cyanotic but may present with signs of heart failure later in infancy. Patients with pulmonary oligemia generally have type I (normally related great arteries); those with pulmonary plethora usually have type II (transposition of the great arteries) and, rarely, type Ic.
The magnitude of pulmonary blood flow without previous surgery largely depends on the degree of pulmonary outflow tract obstruction and patency of the ductus arteriosus. In patients with a type I defect, the obstruction is valvar, subvalvar, or, most frequently, at the VSD level. In patients with a type II defect, the obstruction is either valvar or subvalvar. In patients with a type I defect, if the VSD is large and nonrestrictive without pulmonary stenosis, the pulmonary flow is inversely proportional to the pulmonary-to-systemic vascular resistance ratio. If the ductus is patent or if a surgical systemic-to-pulmonary artery shunt was performed, the pulmonary blood flow is proportional to the size of the natural or surgical aortopulmonary connection.
Left ventricular volume overloading
The left ventricle ejects the entire systemic, coronary, and pulmonary outputs. Therefore, left ventricular volume overloading is present in all patients with tricuspid atresia. [18, 40] The degree of volume overloading increases further if mild or absent pulmonary outflow obstruction is noted or if systemic-to-pulmonary artery shunt was performed. Because normal left ventricular function is critical to a successful Fontan operation, maintenance of normal left ventricular function is essential. Left ventricular function tends to decrease with increasing age, increasing Qp:Qs, and arterial desaturation. [43, 44, 45]
Obstruction of the interatrial communication
Patency of the interatrial communication, usually a patent foramen ovale, is essential for survival. Because the entire systemic venous blood must egress through the interatrial communication, development of interatrial obstruction is not unexpected, but it is rarely clinically significant, especially in the neonate. Right-to-left shunting occurs in late atrial diastole, with augmentation of flow during atrial systole.
Obstruction of the patent foramen ovale is presumed to be present if the mean pressure difference between the atria is more than 5 mm Hg and a tall a wave is present in the right atrial pressure trace. [33] Clinical evaluation may reveal prominent a waves in the jugular venous pulse, presystolic hepatic pulsations, and hepatomegaly. One study suggested that atrial septal aneurysm and an atrial septal defect diameter smaller than 5 mm are associated with an increased risk for developing an atrial septal obstruction. [46]
Changing hemodynamics
Several changes in hemodynamics occur as infants with tricuspid atresia grow. These involve the ductus arteriosus, ASD, and VSD.
Closure of the ductus arteriosus in a neonate with severe pulmonary outflow tract obstruction or atresia results in severe hypoxemia, and the administration of prostaglandin E1 (PGE1) or surgical creation of systemic-to-pulmonary artery shunt is required.
Regarding ASD, restrictive interatrial communication may develop, causing systemic venous congestion. Transcatheter or surgical atrial septostomy may be needed.
Patency of the VSD is essential to maintain intracardiac shunting necessary for patient survival; these VSDs have been named physiologically advantageous VSDs. [27, 47] Functional [48] and partial or complete anatomic [23, 26, 47, 49] closures have been documented. Intermittent functional closure of the VSD results in cyanotic spells in tricuspid atresia. [48] The etiology of such closures has not been identified but is likely similar to that postulated for tetralogy of Fallot.
Closure of a VSD in type I may result in progressive cyanosis, increasing polycythemia, and diminution or disappearance of the murmur of VSD. Both partial and complete closures are reported and require surgical intervention earlier than otherwise anticipated.
Closure of a VSD in type II (transposition) produces subaortic (ie, systemic) outflow obstruction. Partial closures have been reported; however, to the author's knowledge, complete closures have not been documented. Partial closures result in increased left ventricular mass, complicating Fontan operations.
From the author's studies [26, 50] and those of Sauer and Hall, [51] the estimated prevalence of spontaneous VSD closures is 38-48%. [50] This prevalence is similar to that of isolated VSDs. [52, 53] VSD closures are documented in patients aged 1 year to 20 years, with a median of age 1.3 years. These statistics are also similar to those observed in isolated defects.
The most common mechanism of closure is progressive muscular encroachment of margins of the defect with subsequent fibrosis and covering by endocardial proliferation, although other mechanisms of closure seen in isolated VSDs have been observed in tricuspid atresia patients. How such closures are initiated is unknown.
Etiology
The etiology of tricuspid atresia is unknown.
A multifactorial inheritance hypothesis is offered to explain all congenital heart defects, including tricuspid atresia. This hypothesis states that disease results if a predisposed fetus is exposed to a given environmental trigger (to which the fetus is sensitive) during a critical period of cardiac morphogenesis. This genetic and environmental interaction is most likely the pathogenic mechanism for congenital heart defects in general and for tricuspid atresia in particular.
Various risk factors are statistically associated with certain heart defects. However, no specific factors are clearly identified for tricuspid atresia.
Epidemiology
United States data
Although the true incidence of tricuspid atresia is not well defined, the prevalence of tricuspid atresia among congenital heart defects was estimated to be 2.9% in autopsy series and 1.4% in clinical series after extensive review. [54] Given the prevalence of congenital heart defects in 0.8% of live births, tricuspid atresia may be estimated to occur in approximately 1 per 10,000 live births. [54]
International data
Extensive review of the literature indicated no differences in prevalence in tricuspid atresia between the United States and countries on other continents (see the image below), although geographic differences in prevalence for aortic stenosis and coarctation have been documented.
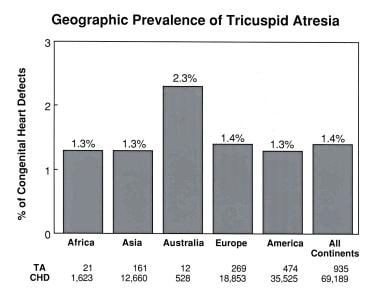 Geographic prevalence of tricuspid atresia by continent. Note that prevalences are similar on all continents except for Australia. This lone exception is thought to be related to the small sample size from Australia. CHD = congenital heart defects; TA = tricuspid atresia. From Rao PS. Demographic features of tricuspid atresia. In: Rao PS, ed, Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:23, with permission.
Geographic prevalence of tricuspid atresia by continent. Note that prevalences are similar on all continents except for Australia. This lone exception is thought to be related to the small sample size from Australia. CHD = congenital heart defects; TA = tricuspid atresia. From Rao PS. Demographic features of tricuspid atresia. In: Rao PS, ed, Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:23, with permission.
Race-related demographics
Although data in the 1950s and early 1960s indicated that the prevalence of congenital heart disease was higher in whites than in blacks, a thorough and appropriate statistical analysis by Mitchell et al suggests that the prevalences of congenital heart disease are similar in whites and blacks (8.3 vs 8.1 per 1000). [55] According to Schriere, the incidences of tricuspid atresia among congenital heart defects in South Africa are 1.2% in whites and 1.4% in African blacks , indicating no racial predilection. [56]
Furthermore, extensive review and tabulation of the prevalences of tricuspid atresia in populations on several continents revealed no difference in prevalences despite different racial compositions on these continents (see the image below). Therefore, no specific racial predilection is noted for tricuspid atresia. [54]
Geographic prevalence of tricuspid atresia by continent. Note that prevalences are similar on all continents except for Australia. This lone exception is thought to be related to the small sample size from Australia. CHD = congenital heart defects; TA = tricuspid atresia. From Rao PS. Demographic features of tricuspid atresia. In: Rao PS, ed, Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:23, with permission.
Sex-related demographics
Some researchers have found a slight male preponderance for tricuspid atresia. An extensive review of 1857 cases revealed that 53% of cases occurred in male individuals and 47% occurred in female individuals. However, these findings were not statistically significant (P >.1), indicating no evidence for sex predilection. [54]
Dick et al suggested that a male preponderance exists only in patients with tricuspid atresia with associated transposition. [33] To test this hypothesis, the authors (1992) evaluated data of patients in whom sex and great-artery relationships were known. In patients without transposition of the great arteries, the prevalences were 54% in male patients and 46% in female patients (P >.1). In patients with transposition of the great arteries, the prevalence was higher in male patients than in female patients (66% vs 34%, P< .05). Therefore, a male preponderance for tricuspid atresia was observed in patients with transposition of the great arteries (type II).
Age-related demographics
Patients with tricuspid atresia present early in life. One half of patients present on the first day of life, two thirds present by the end of the first week, and 80% present by the first month of life. [2, 18, 41] No more than 15% of patients present with symptoms for the first time after 2 months of life.
The magnitude of pulmonary blood flow determines the timing and mode of presentation. Neonates with pulmonary oligemia present early in life with cyanosis, whereas those with pulmonary plethora present slightly later with signs of congestive heart failure, cyanosis, or both, depending on the magnitude of pulmonary flow.
Prognosis
Mortality/Morbidity
Poor prognosis of untreated tricuspid atresia patients is well known; only 10-20% of infants may live through the first year of life.
The image below shows actuarial survival rates from 3 medical centers that Dick and Rosenthal compiled. [41] Considerable early mortality occurs and may be related to hypoxemia, cardiac failure, surgical intervention, or their combination. Surgical palliation to normalize pulmonary blood flow by means of systemic-to-pulmonary artery shunts in neonates with pulmonary oligemia and banding of the pulmonary artery in infants with markedly increased pulmonary flow improves survival rates.
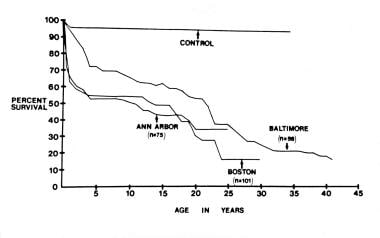 Actuarial survival curves from 3 reported clinical series compiled by Dick and Rosenthal (1992) show a high initial mortality rate in the first year of life, a plateau between the first year and middle of the second decade of life, and a second increase in the mortality rate after the middle of the second decade of life; this second rise is presumably related to impaired left ventricular function. From Dick M and Rosenthal A. The clinical profile of tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. Mt Kisco, NY: Futura Publishing Co; 1982:83, with permission.
Actuarial survival curves from 3 reported clinical series compiled by Dick and Rosenthal (1992) show a high initial mortality rate in the first year of life, a plateau between the first year and middle of the second decade of life, and a second increase in the mortality rate after the middle of the second decade of life; this second rise is presumably related to impaired left ventricular function. From Dick M and Rosenthal A. The clinical profile of tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. Mt Kisco, NY: Futura Publishing Co; 1982:83, with permission.
The availability of PGE1 to keep the ductus open and advances in neonatal care (eg, early identification, safe transport to a tertiary care institution, noninvasive diagnosis by means of echocardiography), anesthesia, and surgical techniques should further decrease the initial mortality rate. [57]
After the early high mortality rate, survival curves become stable and reach a plateau, as shown in the figure below. In patients aged approximately 15 years, a second fall in survival begins and continues through the remaining observation period. Physiologically corrective Fontan procedures may reverse this late mortality. Whether the benefits of Fontan procedure (ie, improving hypoxemia and eliminating left ventricular volume overloading) improve survival rates is not clear. Preliminary data suggest that they do, even after the immediate and late mortality of the surgery itself are accounted for. This potential for improved prognosis means that each patient with tricuspid atresia should undergo aggressive medical and surgical treatment. The natural history after a Fontan operation is shown below.
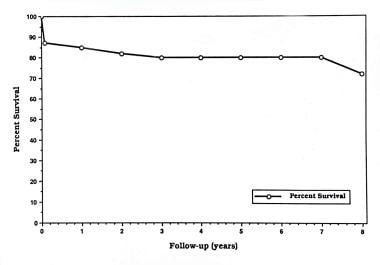 Actuarial survival rate of 100 patients who had tricuspid atresia and who underwent a Fontan operation at the Hospital for Sick Children, Toronto, in 1975-1989. The survival rate 5 years after the operation is 70%. From Freedom RM, et al. The Fontan procedure for patients with tricuspid atresia: long-term follow-up. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:377, with permission.
Actuarial survival rate of 100 patients who had tricuspid atresia and who underwent a Fontan operation at the Hospital for Sick Children, Toronto, in 1975-1989. The survival rate 5 years after the operation is 70%. From Freedom RM, et al. The Fontan procedure for patients with tricuspid atresia: long-term follow-up. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:377, with permission.
Adult patients who had classic Fontan operation have high initial mortality (28%) and high morbidity rates. [58] The latter is related to reoperation (58%) to revise Fontan connections, arrhythmia (56%) and thromboembolic events (25%). Patients with a total cavopulmonary connection appear to have improved survival and decreased morbidity rates, although follow-up of these patients has been relatively short.
The natural history of the component defects (ie, patent ductus arteriosus, ASD and/or patent foramen ovale, and VSD) is described above.
Complications
Development of bacterial endocarditis, brain abscess, and stroke may be considered as complications of the disease itself. Arrhythmias, obstructed venous pathway, and protein-losing enteropathy are some of the complications observed after Fontan surgery.
Patient Education
Tricuspid atresia is a complex cardiac defect requiring multiple and sometimes frequent medical, transcatheter, and surgical interventions. A detailed explanation of the cardiac defects (including pictorial drawings and heart models) and treatment required should be given to the parents at the time of diagnosis and repeated as needed.
For patient education resources, see Heart Health Center as well as Tetralogy of Fallot.
-
Cardiac specimen from a patient with the muscular type of tricuspid atresia. The right atrium was opened by cutting through the right atrial appendage (RAA). Note the dimple (arrow) in the floor of the right atrium with muscle fibers radiating around it. An atrial septal defect (ASD) is also shown. From Rao PS, Levy JM, Nikicicz E, Gilbert-Barness EF. Tricuspid atresia: association with persistent truncus arteriosus. Am Heart J 1991, 122:829, with permission.
-
Systemic arterial saturations. Left ventricular (LV) and aortic (Ao) values are plotted against the pulmonary-to-systemic blood-flow ratio (Qp:Qs). Both type I and type II anatomy are included. Note the curvilinear relationship between the parameters. At low Qp:Qs levels, a slight increase in Qp:Qs produces large increase in systemic oxygen saturation; at high Qp:Qs levels, a further increase does not produce a notable increase in oxygen saturation. The ideal Qp:Qs appears to be 1.5-2.5, which results in oxygen saturations in the low 80s. From Rao PS. Cardiac catheterization in tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co: 1982:153, with permission.
-
Geographic prevalence of tricuspid atresia by continent. Note that prevalences are similar on all continents except for Australia. This lone exception is thought to be related to the small sample size from Australia. CHD = congenital heart defects; TA = tricuspid atresia. From Rao PS. Demographic features of tricuspid atresia. In: Rao PS, ed, Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:23, with permission.
-
Actuarial survival curves from 3 reported clinical series compiled by Dick and Rosenthal (1992) show a high initial mortality rate in the first year of life, a plateau between the first year and middle of the second decade of life, and a second increase in the mortality rate after the middle of the second decade of life; this second rise is presumably related to impaired left ventricular function. From Dick M and Rosenthal A. The clinical profile of tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. Mt Kisco, NY: Futura Publishing Co; 1982:83, with permission.
-
Actuarial survival rate of 100 patients who had tricuspid atresia and who underwent a Fontan operation at the Hospital for Sick Children, Toronto, in 1975-1989. The survival rate 5 years after the operation is 70%. From Freedom RM, et al. The Fontan procedure for patients with tricuspid atresia: long-term follow-up. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:377, with permission.
-
Use of electrocardiographic mean QRS vector (axis) in the frontal plane for the differential diagnosis of a cyanotic newborn with decreased pulmonary blood flow. Associated ventricular hypertrophy patterns (as marked in each quadrant) and decreased right ventricular (RV) forces are also helpful. From Rao PS. Management of neonate with suspected serious heart disease. King Faisal Spec Hosp Med J 1984 (4):209, with permission.
-
Frontal plane mean QRS vector in 308 patients, plotted by anatomic type. Most patients with tricuspid atresia type I (normally related great arteries) have an abnormally superior vector, also called left axis deviation. Only one half of patients with tricuspid atresia type II have an abnormally superior vector. Most patients with tricuspid atresia type III (subtype A) have an inferiorly oriented frontal plane vector. From Rao PS, Kulungara RJ, Boineau JP. Electrovectorcardiographic features of tricuspid atresia. In: Rao PS, ed, Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:141, with permission.
-
Subcostal 4-chamber 2-dimensional echocardiographic view of a neonate with tricuspid atresia shows an enlarged left ventricle (LV), a small right ventricle (RV), and a dense band of echoes where the tricuspid valve echo should be. Atrial and ventricular septal defects and the mitral valve are also seen. Note the attachment of the anterior leaflet of the detectable atrioventricular valve to the left side of the interatrial septum. Reprinted from Rao, Fetal and Neonatal Cardiology, 1990, with permission from Elsevier Science.
-
Selected cineangiographic frames from superior vena caval (SVC) and right atrial (RA) frontal angiography in 2 patients in the frontal view. Note sequential opacification of the left atrium (LA) and left ventricle (LV) without opacification of the right ventricle. The RA on the right, the LA superiorly, and the LV on the left form a nonopacified right ventricular window (arrow). This is a classic appearance of the muscular variety of tricuspid atresia. From Rao PS. Tricuspid atresia: anatomy, imaging, and natural history. In: Brawnwald E, Freedom RM, eds. Atlas of Heart Disease: Congenital Heart Disease. Vol 12. Philadelphia, PA: Current Medicine; 1997:14.1 with permission.
-
Selective superior vena caval (SVC) injection for a 4-chamber projection (hepatoclavicular) shows tricuspid atresia and filling of the left atrium (LA) through a somewhat restrictive atrial septal defect (arrows). Note the retrograde filling of the coronary sinus (CS). RA = right atrium. From Schwartz DC, Rao PS. Angiography in tricuspid atresia. In: Rao PS, ed. Tricuspid Atresia. 2nd ed. Mt Kisco, NY: Futura Publishing Co; 1992:223, with permission.





