Background
Interrupted aortic arch (IAA) is a relatively rare genetic disorder that usually occurs in association with a nonrestrictive ventricular septal defect (VSD) and ductus arteriosus or, less commonly, with a large aortopulmonary window or truncus arteriosus. [1] Although most cases occur in normally connected great arteries, interrupted aortic arch can coexist with any ventriculoarterial alignment and also with severe underdevelopment of one ventricle. The rare cases that involved interrupted aortic arch, aortic valve atresia, and VSD have been complex; two have presented with circle of Willis–dependent coronary blood flow, [2, 3] and two have presented with bilateral ductus in which coronary blood flow depended on the patency of the right ductus arteriosus. [4, 5]
Interrupted aortic arch and complete common atrioventricular canal can be observed in the context of coloboma, heart disease, atresia choanae, retarded growth and development and/or CNS anomalies, genital hypoplasia, and ear anomalies and/or deafness (CHARGE) syndrome, which is usually caused by mutations in CHD7 on chromosome 8q12.1. [6, 7] Approximately 50% of patients with interrupted aortic arch have DiGeorge syndrome; in these cases, the interrupted aortic arch is usually type B, although cases of type A or type C have also been reported. There is considerable phenotypic overlap between CHARGE and DiGeorge syndromes. [8]
Surgical reconstruction of the arch is now relatively straightforward; hence, attention is increasingly focused on the preoperative identification and surgical management of the aortic valve and subaortic stenosis found in approximately one half of cases. [9] Interrupted aortic arch is the first cardiovascular pattern formation anomaly to be demonstrated to have a genetic basis in both mouse and human.
Embryology
Approximately one half of patients with interrupted aortic arch have a hemizygous deletion of a 1.5-3 Mb region of chromosome band 22q11.2, [10, 11] the most common deletion syndrome in humans. Among the 35-50 genes deleted, the T-box gene TBX1 appears to be responsible for most aspects of the DiGeorge phenotype.
In addition, two independent lines of evidence suggest that the etiology of many cases of interrupted aortic arch type A is different from the etiology of interrupted aortic arch type B (see below for definition of types). The variety of associated VSDs is different in the two types of interrupted aortic arch. [12] The prevalence of 22q11.2 hemizygosity is also different; approximately three fourths of patients with interrupted aortic arch type B have the deletion, whereas exceedingly few patients with interrupted aortic arch type A have the deletion.
Anatomy
Interrupted aortic arch has been classified into three types (A, B, and C) based on the site of aortic interruption. In type A interrupted left aortic arch, the arch interruption occurs distal to the origin of the left subclavian artery. In type B interrupted left aortic arch, the interruption occurs distal to the origin of the left common carotid artery. In type C interrupted left aortic arch, the interruption occurs proximal to the origin of the left common carotid artery.
In any of the three types, the right subclavian artery may arise normally or abnormally; the two most common abnormal sites are distal to the left subclavian artery (aberrant right subclavian artery) and from a right ductus arteriosus (isolated right subclavian artery). [13] Type B interruptions account for about two thirds of cases, type A occur in about one third of cases, and type C are present in less than 1% of cases.
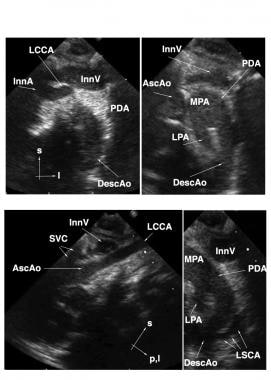 Interrupted Aortic Arch. Suprasternal echocardiographic identification of interrupted aortic arch Type B. Upper left: Frontal (coronal) view showing the takeoffs of the innominate artery (InnA) and left common carotid artery (LCCA). No connection is seen with the distal aorta. Upper right: Slightly more dorsal (posterior) frontal view showing the main pulmonary artery (MPA), large left-sided patent ductus arteriosus (PDA), and left-sided upper descending aorta (DescAo). The ascending aorta (AscAo) is not in discernible continuity with the descending aorta. Lower left: Left oblique view showing the LCCA takeoff and no discernible aortic arch. Lower right: sagittal view showing the origin of the left subclavian artery (LSCA) from the DescAo. Other Abbreviations: InnV= innominate vein; l=left; LPA=left pulmonary artery; p,l = posterior and leftward; s=superior; SVC=superior vena cava.
Interrupted Aortic Arch. Suprasternal echocardiographic identification of interrupted aortic arch Type B. Upper left: Frontal (coronal) view showing the takeoffs of the innominate artery (InnA) and left common carotid artery (LCCA). No connection is seen with the distal aorta. Upper right: Slightly more dorsal (posterior) frontal view showing the main pulmonary artery (MPA), large left-sided patent ductus arteriosus (PDA), and left-sided upper descending aorta (DescAo). The ascending aorta (AscAo) is not in discernible continuity with the descending aorta. Lower left: Left oblique view showing the LCCA takeoff and no discernible aortic arch. Lower right: sagittal view showing the origin of the left subclavian artery (LSCA) from the DescAo. Other Abbreviations: InnV= innominate vein; l=left; LPA=left pulmonary artery; p,l = posterior and leftward; s=superior; SVC=superior vena cava.
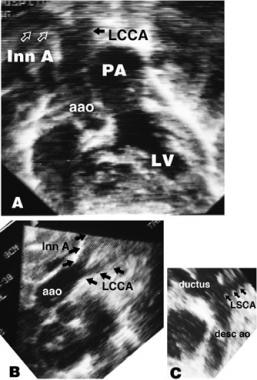 Interrupted Aortic Arch. Section A depicts a subcostal frontal echocardiogram of interrupted aortic arch (IAA) type B with transposition of the great arteries. Section B shows a high parasternal echocardiogram showing that the innominate artery (Inn A) and left common carotid artery (LCCA) arise from the ascending aorta (a ao). In section C, the left subclavian artery (LSCA) arises from the descending aorta (desc ao), which is perfused by the ductus arteriosus.
Interrupted Aortic Arch. Section A depicts a subcostal frontal echocardiogram of interrupted aortic arch (IAA) type B with transposition of the great arteries. Section B shows a high parasternal echocardiogram showing that the innominate artery (Inn A) and left common carotid artery (LCCA) arise from the ascending aorta (a ao). In section C, the left subclavian artery (LSCA) arises from the descending aorta (desc ao), which is perfused by the ductus arteriosus.
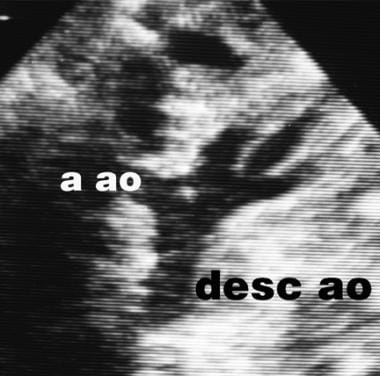 Interrupted Aortic Arch. This is the suprasternal sagittal ultrasonographic view of the patient shown in the previous image. Arch continuity has now been restored by a side-to-side anastomosis. Abbreviations are as follows: a ao = ascending aorta and desc ao = descending aorta.
Interrupted Aortic Arch. This is the suprasternal sagittal ultrasonographic view of the patient shown in the previous image. Arch continuity has now been restored by a side-to-side anastomosis. Abbreviations are as follows: a ao = ascending aorta and desc ao = descending aorta.
Pathophysiology
During fetal development, left ventricular output supplies the arterial circulation proximal to the interruption whereas right ventricular output supplies arterial circulation distal to the interruption via the left ductus arteriosus. Postnatally, this arrangement continues, with the addition of the pulmonary blood flow to the load of the left ventricle. With naturally occurring ductal closure and/or fall in pulmonary vascular resistance after birth, the circulation to the lower part of the body is compromised, resulting in a shocklike state.
Etiology
Abnormalities in any of the cell types involved in formation or patterning the pharyngeal arch arteries (ie, pharyngeal endoderm, [14] pharyngeal mesoderm, endothelium, neural crest) can produce interrupted aortic arch. For example, Tbx1 has a cell-autonomous function in the pharyngeal mesoderm. [15]
More than 25 single-gene mouse knockouts display interrupted aortic arch as a principal phenotype. Mouse mutants displaying interrupted aortic arch are as follows:
-
Foxc2
-
Fgf8 hypomorph
-
Nrp1
-
VEGF-A (knockout of 164 isoform)
-
Cited 2
-
Crkl
-
ET-1 [23]
-
ETA
-
ECE-1
-
FLNA
-
Zic3
-
Dnahc5
-
MRTF-B
-
Sox4
-
AP2α
-
Hoxa1 [24]
-
Gbx2
-
Ltbp1L
-
TGFβ2
-
TGFβRII
-
Bmp4
-
RXR
-
RAR
Although most of these mouse mutants display interrupted aortic arch type B, at least two (Ltbp1L [25] and the myocardin–related transcriptional coactivator Mrtf-B [26] ) can also display interrupted aortic arch type C.
Additional double knockouts or tissue–specific single-gene knockouts with interrupted aortic arch include the following:
-
Chd7+/-Tbx1+/- compound heterozygotes [27]
-
Double null Six1-/-Eya1-/- [28]
-
Second heart field–specific dominant-negative mastermind-like [29]
-
Neural crest–specific knockout of GATA-6 [32]
-
Neural crest–specific knockout of Hdac3 [33]
-
Tbx1 domain–specific knockout of Bmp4 [34]
-
Vascular smooth muscle–specific removal of integrin β1 [35]
-
α5 and αv double null [36]
-
Fgf8 hypomorph; Fgf10 heterozygotes [37]
Approximately 90% of patients with DiGeorge syndrome have deletions within 22q11, which includes TBX1. Rarely, individuals with DiGeorge syndrome have point mutations in TBX1. [38] Patients with DiGeorge syndrome usually have interrupted aortic arch type B, but examples of type A and type C have been reported. Hemizygous deletion of chromosome 1q21.1 [39] can be associated with interrupted aortic arch type A.
Epidemiology
United States data
The incidence is approximately 2 cases per 100,000 live births.
Nearly all patients with interrupted aortic arch present in the first 2 weeks of life when the ductus arteriosus begins to close. Most patients, however, present on the first day of life. Although the vast majority of the IAA cases present in neonates, presentation in childhood, early adulthood, and even in the elderly has been reported. [40, 41, 42, 43, 44, 45, 46] This may be related to persistence of the ductus arteriosus, alternative collateral circulation, and/or a balanced circulation.
Prognosis
In most cases of interrupted aortic arch (IAA), with good surgical repair, the prognosis is excellent.
Morbidity/mortality
Circulatory compromise manifested by metabolic acidosis begins when the ductus arteriosus constricts, thus decreasing flow to the circulation distal to the arch interruption. Prior to this, even severe aortic and subaortic hypoplasia is physiologically masked because of the presence of the VSD. Patients are at risk for severe low output syndrome (ie, cardiogenic shock) because of both the effect of profound metabolic acidosis on cardiac performance and the reduced distal systemic arterial circulation imposed by falling pulmonary vascular resistance.
Complications
Complications in patients with IAA:
-
Persistent subaortic and aortic stenosis [47]
-
Residual ventricular septal defect
-
Narrowing at the site of arch surgery
-
Interrupted Aortic Arch. Suprasternal echocardiographic identification of interrupted aortic arch Type B. Upper left: Frontal (coronal) view showing the takeoffs of the innominate artery (InnA) and left common carotid artery (LCCA). No connection is seen with the distal aorta. Upper right: Slightly more dorsal (posterior) frontal view showing the main pulmonary artery (MPA), large left-sided patent ductus arteriosus (PDA), and left-sided upper descending aorta (DescAo). The ascending aorta (AscAo) is not in discernible continuity with the descending aorta. Lower left: Left oblique view showing the LCCA takeoff and no discernible aortic arch. Lower right: sagittal view showing the origin of the left subclavian artery (LSCA) from the DescAo. Other Abbreviations: InnV= innominate vein; l=left; LPA=left pulmonary artery; p,l = posterior and leftward; s=superior; SVC=superior vena cava.
-
Interrupted Aortic Arch. Section A depicts a subcostal frontal echocardiogram of interrupted aortic arch (IAA) type B with transposition of the great arteries. Section B shows a high parasternal echocardiogram showing that the innominate artery (Inn A) and left common carotid artery (LCCA) arise from the ascending aorta (a ao). In section C, the left subclavian artery (LSCA) arises from the descending aorta (desc ao), which is perfused by the ductus arteriosus.
-
Interrupted Aortic Arch. This is the suprasternal sagittal ultrasonographic view of the patient shown in the previous image. Arch continuity has now been restored by a side-to-side anastomosis. Abbreviations are as follows: a ao = ascending aorta and desc ao = descending aorta.





