Background
X-linked immunodeficiency with hyper–immunoglobulin M (XHIGM or HIGM1) is a rare form of primary immunodeficiency disease caused by mutations in the gene that codes for CD40 ligand (CD40L, also known as CD154 or TNFSF5 or gp39). CD40L is expressed on activated T lymphocytes and is necessary for T cells to induce B cells to undergo immunoglobulin (Ig) class-switching from immunoglobulin M (IgM) to immunoglobulin G (IgG), immunoglobulin A (IgA), and immunoglobulin E (IgE). [1]
Thus, patients with XHIGM have markedly reduced levels of IgG, IgA, and IgE but have normal or elevated levels of IgM. Because CD40L is required in the functional maturation of T lymphocytes, dendritic cells, and macrophages, patients with XHIGM also have a variable defect in T-lymphocyte, dendritic cells, and macrophage effector function. Clinically, patients with XHIGM have increased susceptibility to infection with a wide variety of bacteria, viruses, fungi, and parasites. In addition, they are at increased risk for developing autoimmune disorders and malignancies.
Since the first description of patients with XHIGM by Rosen et al in 1961, several other genetic defects have been reported that are associated with defective Ig class-switch recombination (CSR). In 1974, a World Health Organization (WHO) working party named the syndrome immunoglobulin deficiency with increased IgM (hyper-IgM syndrome [HIGM1]). [2] The most common form of HIGM is XHIGM (or HIGM1) and is inherited as an X-linked recessive (XR) trait. Another XR form of the syndrome is associated with hypohidrotic ectodermal dysplasia. In addition, several autosomal recessive forms (AR) and an autosomal dominant form of HIGM have been reported (see Differentials). In 2015, the International Union of Immunology Societies (IUIS) Phenotypic Classification for Primary Immundeficiencies named it CD40 ligand deficiency and classified XHIGM under combined immunodeficiency without T cell lymphopenia. [35]
Pathophysiology
Humoral immunity, or antibody-mediated immune responses, plays a central role in defense against extracellular pathogens and some viruses. Humoral immunity depends on the generation of exquisite specificity and diversity of Igs. During the primary antibody response, B cells in the bone marrow produce IgM and immunoglobulin D (IgD) antibodies of low avidity. This process is largely antigen-independent.
Once IgM B cells are engaged with antigens, B cells start the secondary antibody repertoire generation by undergoing 2 genetic alterations to improve specificity and avidity of the antibody to specific microorganisms.
The first step is generation of Ig diversity by recombination of Ig heavy chain, known as class-switch recombination (CSR), switching from IgM to IgG, IgA, or IgE.
The second step is somatic hypermutation (SHM) and involves the introduction of point mutations in the V regions (antigen-binding sites) of the Ig genes, resulting in an expansion of the antibody repertoire to generate high-affinity antigen-specific antibodies. The secondary antibody repertoire generation is antigen and T-cell dependent and occurs in peripheral lymphoid organs, mainly through the interaction between CD40L (CD154), expressed on activated CD4+ T cells, and CD40, expressed on B cells (see image below).
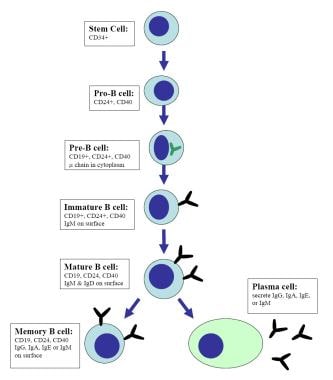 During the primary antibody response, B cells in the bone marrow produce immunoglobulin M (IgM) and immunoglobulin D (IgD) antibodies of low avidity. This process occurs largely in an antigen-independent way (pro-B cells, pre-B cells). Once IgM B cells are engaged with antigens, B cells start the secondary antibody repertoire generation by undergoing 2 genetic alterations; class-switch recombination (switching from IgM to IgG, IgA, or IgE) and somatic hypermutation (introduction of point mutations in the V regions of the Ig genes, the antigen-biding sites, resulting in an expansion of the antibody repertoire to generate high-affinity antigen-specific antibodies). The secondary antibody repertoire generation is antigen and T-cell dependent and occurs in peripheral lymphoid organs, mainly through the interaction between CD40L (CD154) expressed on activated CD4+ T cells and CD40 expressed on B cells.
During the primary antibody response, B cells in the bone marrow produce immunoglobulin M (IgM) and immunoglobulin D (IgD) antibodies of low avidity. This process occurs largely in an antigen-independent way (pro-B cells, pre-B cells). Once IgM B cells are engaged with antigens, B cells start the secondary antibody repertoire generation by undergoing 2 genetic alterations; class-switch recombination (switching from IgM to IgG, IgA, or IgE) and somatic hypermutation (introduction of point mutations in the V regions of the Ig genes, the antigen-biding sites, resulting in an expansion of the antibody repertoire to generate high-affinity antigen-specific antibodies). The secondary antibody repertoire generation is antigen and T-cell dependent and occurs in peripheral lymphoid organs, mainly through the interaction between CD40L (CD154) expressed on activated CD4+ T cells and CD40 expressed on B cells.
B cells of patients with XHIGM are intrinsically normal, in that they can be induced to proliferate and undergo CSR upon in vitro activation by CD40 agonists and appropriate cytokines. CD40 activation is also necessary for B cells to act as antigen-presenting cells, further enhancing the adaptive (acquired) immune response of T cells and other cells. Although B cells mature to express CD19 and surface immunoglobulins in the absence of CD40L on T cells, differentiation to plasma cells does not occur.
Because CD40 is also expressed on monocytes and dendritic cells, impaired CD40L expression leads to defective T-cell interactions with monocytes and dendritic cells, resulting in abnormal cell-mediated immune function and increased susceptibility to opportunistic infections, fungal infection, malignancy, and autoimmune diseases (28).
Neutropenia is also a common feature of XHIGM and may result from a defective, stress-induced, CD40-dependent granulopoiesis as myeloid progenitors express CD40 molecules. CD40L and CD40 are widely expressed on hematopoietic cells, and CD40 triggering on stromal cells enhances the expression of granulopoiesis growth factors, such as granulocyte-colony-stimulating factor (G-CSF) and granulocyte/monocyte-colony-stimulating factor (GM-CSF). Disruption of the CD40L/CD40-signaling pathway can lead to neutropenia. [3]
Increased incidence of autoimmune disorders have been reported among patients with HIGM syndrome. Furthermore CD40L-CD40 interactions may play an important role in T-regulatory (T-reg) cells that are required for the establishment and maintenance of immune tolerance. Patients with CD40L deficiency displayed low numbers of T-reg cells and defects in B-cell tolerance.
Epidemiology
Frequency
United States
In 2003, the US XHIGM registry reported that the minimal incidence rate of XHIGM was approximately 1 in 1,000,000 live births from 1984-1993. [4] This may be an underestimation because not all physicians in the United States participated in the registry.
International
Establishing reasonable estimates of XHIGM incidence has been difficult because most primary immunodeficiency disease registries combine data regarding XHIGM with data regarding genetic defects with resultant hyper-IgM. All forms of HIGM constitute 0.3-2.9% of all patients with primary immunodeficiencies in Europe, Asia, and South America. One registry in Spain reported that the incidence rate of all forms of HIGM is 1 per 20 million live births. XHIGM represents about 65-70% of all HIGMs. [5]
Mortality/Morbidity
A retrospective study of the Registry of the European Society for Immune Deficiency of 56 affected males showed a 20% survival rate in individuals aged 25 years. [6] The US XHIGM Registry reported that 11 of 61 surviving patients were aged 20 years or older. [4]
The leading cause of death was pneumonia, encephalitis, or malignancy. Other patients died of liver failure secondary to sclerosing cholangitis and cirrhosis.
Major causes of morbidity include infection with bacteria, fungi, or viruses and opportunistic infections such as those involving Pneumocystis carinii. The respiratory (sinopulmonary) system, CNS, hepatobiliary system, and skin are commonly affected. Chronic diarrhea with or without infection has been frequently reported. Neutropenia, anemia, and thrombocytopenia are common. An increased risk of GI tract malignancies has been reported. Morbidity due to infection has markedly improved with the advent of intravenous immunoglobulin (IVIG) replacement therapy and better recognition of Pneumocystis jiroveci pneumonia and early initiation of Pneumocystis prophylaxis.
Race
Studies are inadequate to provide ethnic data regarding XHIGM incidence. The US XHIGM Registry reported the racial background of 75 patients. [4] Fifty two of the patients were white, 12 were black, 9 were Asian, 1 was both black and Asian, and 1 was white and Asian.
Sex
CD40L deficiency affects males because it is an inherited in XR trait. Female carriers, even with extreme lyonization, have been immunocompetent and without clinical illness. Females with hypogammaglobulinemia and high IgM levels should be tested for gene mutations that affect other forms of HIGM.
Age
Most patients are diagnosed before age 4 years. Over one half of patients develop symptoms of immunodeficiency by age 1 year, and nearly all develop symptoms by age 4 years.
-
During the primary antibody response, B cells in the bone marrow produce immunoglobulin M (IgM) and immunoglobulin D (IgD) antibodies of low avidity. This process occurs largely in an antigen-independent way (pro-B cells, pre-B cells). Once IgM B cells are engaged with antigens, B cells start the secondary antibody repertoire generation by undergoing 2 genetic alterations; class-switch recombination (switching from IgM to IgG, IgA, or IgE) and somatic hypermutation (introduction of point mutations in the V regions of the Ig genes, the antigen-biding sites, resulting in an expansion of the antibody repertoire to generate high-affinity antigen-specific antibodies). The secondary antibody repertoire generation is antigen and T-cell dependent and occurs in peripheral lymphoid organs, mainly through the interaction between CD40L (CD154) expressed on activated CD4+ T cells and CD40 expressed on B cells.
-
Chest radiograph of a 22-year-old patient with X-linked hyper-IgM syndrome.
-
Chest radiograph (laterval view) of 22-year-old patient with X-linked hyper-IgM syndrome.
-
Chest CT scan of a 22-year-old patient with X-linked hyper-IgM syndrome.
Tables
|
XHIGM |
CD40 defect |
EDA-ID |
AR-AID |
AID- Cter |
AID-Δ C |
UNG defect |
CSR defect- upstream from DNA cleavage |
CSR defect-downstream from DNA cleavage |
Defect |
CD40LG |
CD40 |
NEMO |
AICDA |
AICDA |
AICDA |
UNG |
Unknown |
Unknown |
Inheritance |
XL |
AR |
XL |
AR |
AR |
AD |
AR |
AR |
AR |
Lymphadenopathy |
- |
- |
- |
++ |
++ |
++ |
+ |
+ |
+ |
Opportunistic Infection |
+ |
+ |
- |
- |
- |
- |
- |
- |
- |
Autoimmunity |
± |
± |
+ |
+ |
+ |
+ |
- |
- |
+ |
Serum IgM |
N or ↑ |
N or ↑ |
N or ↑ |
↑ ↑ |
↑ ↑ |
↑ |
↑ ↑ |
N or ↑ |
N or ↑ |
CD40-induced CSR |
N |
UD |
Variable |
UD |
UD |
UD |
UD |
UD |
UD |
SHM |
↓ |
↓ |
Variable |
↓ ↓ |
N |
N |
N but biased |
N |
N |
Brand (Manufacturer) |
Virus Inactivation process |
pH; Additives * |
Osmolality (mOsm/kg) |
Parenteral Form & Final Concentrations |
IgA Content ( µg/ml) |
IV or SC |
Bivigam (Biotest Pharmaceuticals) |
Cold ethanol fractionation, solvent/detergent, nanofiltration |
4.0-4.6; glycine, polysorbate 80 |
Unspecified |
Liquid 10% |
< 200 µg/mL |
IV |
Flebogamma DIF(Grifols) |
Pasteurization, solvent/detergent, nanofiltration, fractionation, low pH treatment |
5.0-6.0; D-Sorbitol |
240-370 |
Liquid 5%, 10% |
< 50µg/mL in a 5% solution, < 100µg/mL in a 10% solution |
IV |
Gammagard S/D Low IgA (Baxter) |
Cold ethanol fractionation, solvent/detergent |
6.4-7.2; Albumin, glycine, glucose, PEG, tri-n-butyl phosphate, octoxynol, polysorbate 80 |
5%=636; 10%=250 |
Lyophilized powder 5%, 10% |
< 1µg/mL in a 5% solution |
IV |
Gammaplex (Bio Products Laboratory) |
Solvent/detergent, nanofiltraion, low pH incubation |
4.8-5.1; D- sorbitol, glycine, polysorbate 80 |
420-500 |
Liquid 5% |
< 10 µg/mL |
IV |
Octagam (Octapharma) |
Cold ethanol fractionation, solvent/detergent, pH4 treatment |
5.1-6.0; maltose |
310-380 |
Liquid 5% |
< 200 µg/mL |
IV |
Privigen (CSL Behring) |
pH 4 incubation, nanofiltration, depth filtration |
4.6-5.0; L-proline |
240-440 |
Liquid 10% |
< 25 µg/mL |
IV |
Gammagard Liquid (Baxter) |
Solvent/detergent, nanofiltration, low pH incubation at elevated temp |
4.6-5.1; glycine |
240-300 |
Liquid 10% |
37 µg/mL |
IV or Subcutaneous |
Gamunex-C (Grifolis) |
Caprylate precipitation, depth filtration, chromatography, pH 4 incubation |
4-4.5; glycine |
258 |
Liquid 10% |
46 µg/mL |
IV or Subcutaneous |
Gammaked (Kedrion Biopharma) |
Caprylate precipitation, depth filtration, chromatography, low pH incubation |
4.0-4.5; glycine |
258 |
Liquid 10% |
46 µg/mL |
IV or Subcutaneous |
Cuvitru (Shire) |
Fractionation, SD treatment, nanofiltration, low pH treatment |
4.6-5.1; glycine |
280-292 |
Liquid 20% |
80 µg/mL |
Subcutaneous |
Hizentra (CSL Behring) |
Cold alcohol fractionation, octanoic acid fractionation, and anion exchange chromatography |
4.6-5.2; L-proline, polysorbate 80 |
380 |
Liquid 20% |
< 50 µg/mL |
Subcutaneous |
HyQvia (Baxter Healthcare) |
Solvent/detergent, nanofiltration, low pH incubation |
4.6-5.1; recombinant human hyaluronidase (pH 7.4) |
240-300; (290-350) |
Liquid 10% |
37 µg/mL |
Subcutaneous |
Xembify (Grifolis) |
Cold ethanol fractionation, caprylate precipitation and filtration, and anion-exchange chromatography, low pH incubation | 4.1-4.8 polysorbate 80 |
280-404 | Liquid 20% | Subcutaneous |




