Practice Essentials
Xanthinuria is a descriptive term for excess urinary excretion of the purine base xanthine. Two inherited forms of xanthinuria principally result from a deficiency of the enzyme xanthine dehydrogenase, which is the enzyme responsible for degrading hypoxanthine and xanthine to uric acid. Deficiency of xanthine dehydrogenase results in plasma accumulation and excess urinary excretion of the highly insoluble xanthine, which may lead to arthropathy, myopathy, crystal nephropathy, urolithiasis, or renal failure. Hypoxanthine does not accumulate to an appreciable degree because it is recycled through a salvage pathway by the enzyme hypoxanthine guanine phosphoribosyltransferase (HGPRT). Xanthine continues to accumulate, despite the recycling of hypoxanthine, because of the metabolism of guanine to xanthine by the enzyme guanase (see image below).
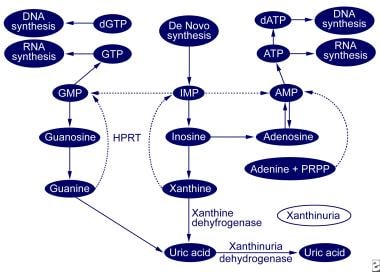 Purine metabolic pathway.
Purine metabolic pathway.
Background
Classic xanthinuria
Classic xanthinuria is one form of xanthinuria that is divided into 2 types based on the enzyme deficiency. Both types are inherited in an autosomal recessive manner.
-
Classic xanthinuria type I is the result of an isolated deficiency of xanthine dehydrogenase.
-
Type II xanthinuria is characterized by a deficiency of xanthine dehydrogenase and a related enzyme, aldehyde oxidase.
The distinction between the 2 types is based on the ability or inability to oxidize allopurinol, a substrate for xanthine dehydrogenase and aldehyde oxidase. Allopurinol is oxidized to oxypurinol by the normal function of aldehyde oxidase in patients with type I; however, allopurinol is not converted in patients with type II, who lack aldehyde oxidase activity. Other substrates that are oxidized by aldehyde oxidase, such as pyrazinamide and N -methylnicotinamide, can be used to distinguish between types I and II.
Molybdenum cofactor deficiency
The other inherited form of xanthinuria, termed molybdenum cofactor deficiency, presents in the neonatal period with microcephaly, hyperreflexia, and other CNS manifestations. Other reported manifestations include severe metabolic acidosis and intracranial hemorrhage. This condition is inherited recessively and is caused by a congenital defect of a molybdenum-containing cofactor essential for the function of 3 distinct enzymes (ie, xanthine dehydrogenase, aldehyde oxidase, sulfite oxidase). This defect is caused by the mutation of molybdenum cofactor genes (MOCS1 or MOCS2). Xanthinuria is only a marker in this setting because (1) the clinical presentation is overshadowed by neurologic manifestations and (2) death in the first year of life is caused by the deficiency of sulfite oxidase, which is the final step in cysteine metabolism. [1]
A study reported on the outcome of the complete first cohort of patients receiving substitution treatment with cyclic pyranopterin monophosphate (cPMP), a biosynthetic precursor of the cofactor, to treat molybdenum cofactor deficiency (MoCD). The study concluded that cPMP substitution is the first effective therapy for patients with MoCD type A and has a favorable safety profile. [2]
Iatrogenic xanthinuria
Iatrogenic xanthinuria can occur during allopurinol therapy, which is used to reduce urine uric acid excretion in conditions with endogenous overproduction of uric acid. Inhibition of xanthine dehydrogenase by allopurinol may lead to accumulation and urinary excretion of xanthine. Patients with Lesch-Nyhan syndrome or patients with partial HGPRT deficiency have developed xanthine nephropathy, acute kidney failure, and stones following treatment with allopurinol. [3] A few incidents of xanthine nephropathy and renal failure have been reported in patients treated with allopurinol during chemotherapy for malignancy. The latter occurred either when large doses of allopurinol were used or during aggressive therapy for a large tumor cell burden with concomitant allopurinol therapy.
This discussion focuses on the classic and iatrogenic forms of xanthinuria in children.
Pathophysiology
The primary organs affected in xanthinuria are the kidney and, to a lesser extent, skeletal muscle and joints. Kidney complications are initiated by the formation of xanthine crystals in the tubules, leading to parenchymal deposition and/or radiolucent stone formation. Xanthine's high rate of renal clearance and low solubility in urine creates an environment in the urine favoring crystallization. Thus, patients with volume depletion who have xanthinuria are at particular risk of forming xanthine crystals. Irritation of the tubular epithelium by xanthine crystals results in hematuria, whereas renal tissue deposits induce an inflammatory reaction and consequent interstitial nephritis. Urolithiasis is the most common clinical manifestation of the xanthinuric states. Further renal complications include acute and chronic renal failure and even end-stage renal disease.
Myopathy and arthropathy are rare clinical manifestations of xanthinuria that have been described in older patients. Clinical manifestations of the myopathy (eg, muscle cramps, muscle pain, muscle stiffness) are believed to be the result of long-term accumulation of xanthine and hypoxanthine crystals in the muscle; this has been demonstrated in skeletal muscle biopsies in a few symptomatic patients. A form of myopathy has been described in one patient following vigorous exercise, which led to the postulate that heavy muscle use leads to an intracellular acid environment favoring xanthine and hypoxanthine crystal formation and deposition in muscle tissue. Hypoxanthine serum levels are also increased after vigorous exercise (eg, distance running) in healthy subjects and in patients with xanthinuria.
Arthropathy induced by xanthine crystal deposition has been demonstrated in animals. Although not clearly demonstrated in humans, arthritis and arthralgia are believed to be secondary to xanthine crystal deposition in the joints.
Etiology
Genetic causes
Classic xanthinuria types I and II are autosomal recessive inherited conditions that result in dysfunction of the enzyme xanthine dehydrogenase. Xanthine dehydrogenase catalyzes 2 reactions, conversion of hypoxanthine to xanthine and conversion of xanthine to uric acid.
The accumulation of xanthine is caused by the catabolism of guanine to xanthine by guanase and the lack of a salvage pathway for xanthine. Hypoxanthine does not accumulate appreciably because it is efficiently metabolized through a salvage pathway.
Iatrogenic causes
Allopurinol is administered to block xanthine dehydrogenase and prevent uric acid overproduction, which leads to the accumulation of xanthine. Rarely, in the setting of aggressive chemotherapy with rapid tumor lysis and allopurinol therapy, patients can develop complications of renal failure from xanthine crystal nephropathy. Volume depletion may also be involved.
In complete HGPRT deficiency (ie, Lesch-Nyhan syndrome) or in partial deficiency of HGPRT, an overproduction of uric acid occurs. Allopurinol is administered to reduce uric acid production, and this leads to xanthine and hypoxanthine accumulation. Hypoxanthine accumulates because HGPRT is the enzyme for the hypoxanthine salvage pathway.
Epidemiology
United States data
True incidence of classic xanthinuria is unknown because it is rarely reported. Surveys suggest a population incidence of 1 case per 6,000 population to 1 case per 69,000 population. Distribution of patients with type I and type II is approximately equal. The incidence of iatrogenic xanthinuria is unknown.
International data
Most reported cases are from Mediterranean and Middle Eastern countries.
Race-, sex-, and age-related demographics
Xanthinuria or xanthine dehydrogenase deficiency is reported in diverse ethnicities, although most reported incidents occur in Mediterranean and Middle Eastern countries. Consanguinity and an arid climate appear to have a significant role in the higher incidence in these populations.
Classic xanthinuria is more common in males than in females.
Nephrotoxicity from classic xanthinuria can occur at any age, although more than one half of the incidents of urolithiasis occur in children younger than 10 years. Myopathy and arthropathy occur more often in older patients with xanthinuria.
Prognosis
The prognosis depends on the degree of renal injury from crystal nephropathy, urinary obstruction, and/or pyelonephritis.
Morbidity/mortality
Although the death rate is unknown and unexpected, death can result as a complication of unrecognized or untreated renal failure. Nearly 40% of patients with classic xanthinuria present with symptoms related to urolithiasis (eg, hematuria, renal colic, urinary tract infection, acute renal failure).
Complications
Complications of xanthinuria may include the following:
-
Urolithiasis
-
Crystal nephropathy
-
Renal failure
-
Obstructive uropathy
-
Urinary tract infection
-
Hematuria
-
Myopathy
-
Arthropathy
-
Arthritis
Patient Education
Advise patients regarding the importance of the following:
-
Maintaining a dilute urine
-
Avoiding dehydration
-
Intervening early for conditions that may lead to dehydration
-
Avoiding high-purine foods
-
Providing proper home therapy for renal colic
-
Purine metabolic pathway.


