Practice Essentials
Wilms tumor, or nephroblastoma, is the most common childhood abdominal malignancy. The median age at diagnosis of this kidney tumor (see the image below) is approximately 3.5 years. With current multimodality therapy, approximately 80-90% of children with a diagnosis of Wilms tumor survive. [1]
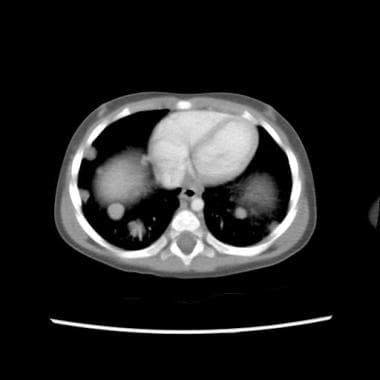 CT scan of child with a stage IV Wilms tumor with favorable histology. Note the bilateral pulmonary metastases.
CT scan of child with a stage IV Wilms tumor with favorable histology. Note the bilateral pulmonary metastases.
See Wilms Tumor: A Pediatric Oncology Success Story, a Critical Images slideshow, to help identify the clinical features, staging evaluation, prognostic factors, and therapeutic options for this disease.
Signs and symptoms
Clinical findings include the following:
-
Asymptomatic abdominal mass (in 80% of children at presentation)
-
Abdominal pain or hematuria (25%)
-
Urinary tract infection and varicocele (less common)
-
Hypertension, gross hematuria, and fever (5-30%)
-
Hypotension, anemia, and fever (from hemorrhage into the tumor; uncommon)
-
Respiratory symptoms related to lung metastases (in patients with advanced disease; rare)
See Presentation for more detail.
Diagnosis
The following laboratory studies are indicated:
-
Complete blood cell (CBC) count
-
Chemistry profile - Including kidney function tests and routine measurements of electrolytes and calcium
-
Urinalysis
-
Coagulation studies
-
Cytogenetics studies, including 1p and 16q deletion
Imaging studies are as follows:
-
Renal ultrasonography (often the initial study)
-
Four-field chest radiography (to detect lung metastases)
-
Abdominal and chest computed tomography (CT)
-
Abdominal magnetic resonance imaging (MRI)
Histologic confirmation in North America is with immediate nephrectomy, with exploration of the contralateral kidney to ensure that the disease is unilateral, and lymph node biopsy sampling for staging purposes. Immediate nephrectomy is not performed in patients with bilateral disease at presentation.
Approximately 90% of all Wilms tumors have favorable histology; however, 3-7% of Wilms tumors are characterized by anaplastic changes. If these changes are present diffusely throughout the tumor, they are predictive of a poor outcome. Wilms tumors with anaplastic changes have unfavorable histology.
See Workup for more detail.
Management
The usual approach is surgery (nephrectomy) followed by chemotherapy, with or without postoperative radiotherapy. Chemotherapy regimens typically comprise vincristine and dactinomycin; doxorubicin and then cyclophosphamide and etoposide are added for increasingly high-risk disease. Children with loss of heterozygosity at 1p and 16q receive more aggressive chemotherapy.
See Treatment and Medication for more detail.
Background
The most common childhood abdominal malignancy is Wilms tumor, or nephroblastoma. Over the past 5 decades, the multidisciplinary approach to this kidney tumor has become an example for successful cancer treatment (see the images below). (See Treatment and Medication.)
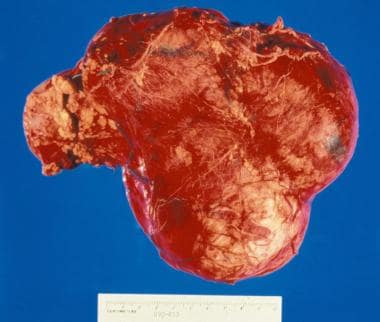 Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side.
Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side.
At present, survival rates of children with Wilms tumor are approximately 80-90%. This is in contrast to the rate 50 years ago, when only 10% of children survived. The addition of radiation therapy to surgery alone improved survival rates to approximately 40%. With the use of chemotherapy, survival rates climbed to their current values. (See Prognosis.)
The National Wilms Tumor Study Group (NWTSG) and the International Society of Pediatric Oncology (SIOP) have identified several chemotherapeutic agents through clinical trials. When used together, these agents lead to a cure in most children with this renal tumor. In addition, the guidelines for surgical treatment and the role of radiation therapy are better defined now than ever before. (See Treatment and Medication.)
With the great improvement in survival rates, therapeutic trials came to focus on limiting treatment-related toxicity. [2] Understanding of the molecular mechanisms that contribute to the development of Wilms tumor has also greatly increased, making Wilms tumorigenesis a model for the understanding of the development of other tumors. (See Etiology.)
Children’s Oncology Group staging of Wilms tumors
Stage I tumors have the following characteristics:
-
The tumor is limited to kidney and is completely resected.
-
The renal capsule is intact.
-
The tumor was not ruptured or biopsied prior to removal.
-
The vessels of the renal sinus are not involved.
-
No evidence of tumor is present at or beyond the margins of resection.
Stage II tumors have the following characteristics:
-
The tumor is completely resected.
-
No evidence of tumor at or beyond the margins of resection is noted.
-
The tumor extends beyond the kidney (penetration of renal capsule, involvement of renal sinus).
Stage III tumors have the following characteristics:
-
A residual, nonhematogenous tumor is present following surgery and is confined to the abdomen.
-
Positive lymph nodes in the abdomen or pelvis are noted.
-
Penetration through the peritoneal surface is observed.
-
Peritoneal implants are present.
-
Gross or microscopic tumor remains postoperatively, including positive margins of resection.
-
Tumor spillage is noted, including biopsy.
-
The tumor is treated with preoperative chemotherapy.
-
The tumor is removed in more than 1 piece.
Stage IV tumors are characterized by the following:
-
Hematogenous metastases (eg, lung, liver, bone, brain) or lymph node metastases beyond the abdomen or pelvis are noted.
Stage V tumors are characterized by the following:
-
Bilateral renal involvement by the tumor is present at diagnosis.
Etiology
Wilms tumor is thought to be caused by alterations of genes responsible for normal genitourinary development. Examples of common congenital anomalies associated with Wilms tumor are cryptorchidism, a double collecting system, horseshoe kidney, and hypospadias. Environmental exposures, although considered, seem relatively unlikely to play a role.
WT1 gene
In the early 1970s, Knudson and Strong proposed a genetic model for the development of Wilms tumor. [3] WT1, the first Wilms tumor suppressor gene at chromosomal band 11p13, was identified as a direct result of the study of children with Wilms tumor who also had aniridia, genitourinary anomalies, and intellectual disability (WAGR syndrome). [4]
Karyotypic analysis revealed constitutional deletions within the short arm of 1 copy of chromosome 11. The 11p13 locus was subsequently demonstrated to encompass numerous contiguous genes, including the aniridia gene PAX6 and the Wilms tumor suppressor gene WT1, which was cloned in 1990. WT1 encodes a transcription factor critical to normal renal and gonadal development.
Characterization of this novel tumor suppressor gene has provided insight into the mechanisms underlying normal kidney development and Wilms tumorigenesis. The WT1 gene is the specific target of mutations and deletions in a subset of patients with sporadic Wilms tumors, as well as in the germline of some children (eg, those with Denys-Drash syndrome) with a genetic predisposition to develop this cancer. [5]
Additional genetic loci
A second gene that predisposes individuals to develop the Wilms tumor has been identified (but has not yet been cloned) telomeric of WT1, at 11p15. This locus was proposed on the basis of studies in patients with both Wilms tumor and Beckwith-Wiedemann syndrome (BWS), another congenital Wilms-tumor predisposition syndrome linked to chromosomal band 11p15. [4]
BWS is an overgrowth syndrome characterized by visceromegaly, macroglossia, and hyperinsulinemic hypoglycemia. In addition, patients with BWS are predisposed to have several embryonal neoplasms, including Wilms tumor. Thus far, a few candidate loci for Wilms tumor and BWS have been proposed. These loci include the insulinlike growth factor II gene (IGFII), H19 (for an untranslated ribonucleic acid [RNA]), and that encoding for p57kip2.
Results of linkage analyses in large pedigrees with familial transmission of susceptibility to the Wilms tumor suggest the existence of additional genetic loci.
Finally, loci at 16q, 1p, 7p, and 17p have also been implicated in the biology of Wilms tumor, although these loci do not seem to predispose individuals to develop a Wilms tumor. Instead, they seem to be associated with the phenotype or the outcome. [2, 6]
Epidemiology
United States data
Wilms tumor affects approximately 10 children and adolescents per 1 million before age 15 years. Therefore, it accounts for 6-7% of all childhood cancers in North America. As a result, about 450-500 new cases are diagnosed each year on this continent. In 5-10% of patients, both kidneys are affected at the same time (synchronous bilateral Wilms tumor) or one after the other (metachronous bilateral Wilms tumor). [7]
International data
Wilms tumor appears to be relatively more common in Africa and least common in East Asia. [7] The incidence in Europe is similar to that reported in North America.
Low-income countries have a higher median incidence of Wilms tumor (9.8 age-standardized rate [ASR] per million) than high-income (8.6 ASR per million) and middle-income countries (6.1 ASR per million). [8]
Race-, sex-, and age-related demographics
Wilms tumor is relatively more common in blacks than in whites and is rare in East Asians. Estimates suggest 6-9 cases per million person years in whites, 3-4 cases per million person years in East Asians, and more than 10 cases per million person years among black populations. [7]
Among patients with unilateral Wilms tumor enrolled in all NWTSG protocols, the male-to-female ratio was 0.92:1. For patients with bilateral disease, the male-to-female ratio was 0.60:1.
The median age at diagnosis of Wilms tumor is approximately 3.5 years. The median age is highest for patients with unilateral unicentric disease (36.1 mo) and lowest for those with synchronous bilateral Wilms tumors (25.5 mo). [7]
A study by Heck et al looked to determine whether the risk of childhood cancers among Hispanic children varies by maternal birthplace. The study on more than 4 million children of non-Hispanic white mothers, more than 2 million children of US-born Hispanic mothers, and 4 million children of non–US born Hispanic mothers from 1983 to 2011 found that for certain cancer types, such as glioma and astrocytoma, solid neuroblastoma, and Wilms tumor of the kidney, children of non–US born Hispanic mothers had the lowest risk. [9]
Prognosis
Approximately 80-90% of children with a diagnosis of Wilms tumor survive with current multimodality therapy. [10] Patients who have tumors with favorable histology have an overall survival rate of at least 80% at 4 years after the initial diagnosis, even in patients with stage IV disease.
The 4-year relapse-free and overall survival rates in patients with favorable-histology Wilms tumor are shown in Table 1.
Table 1. Survival Rates in Patients with Favorable-Histology Wilms Tumor (Open Table in a new window)
Stage |
Relapse-Free Survival, % |
Overall Survival, % |
I |
92 |
98 |
II |
85 |
96 |
III |
90 |
95 |
IV |
80 |
90 |
Patients with synchronous bilateral tumors have a 70-80% survival rate, [11, 12] whereas those with metachronous tumors have a 45-50% survival rate. [13]
Patients with anaplastic Wilms tumor have a worse prognosis compared with favorable histology Wilms tumor; the 4-year overall survival rates are 83%, 83%, 65% and 33% for stages I, II, III, and IV, respectively. [14]
Children with a loss of heterozygosity at 1p and 16q have a worse prognosis than do children without this heterozygosity loss. [6]
The prognosis for patients who have a relapse is not as good as it is for those with a newly diagnosed Wilms tumor, with 40-80% of relapse patients expected to survive after salvage therapy. Patients who relapse after having received vincristine and actinomycin D have a better survival with relapse treatment compared with those who initially received vincristine, actinomycin D, and doxorubicin. [15, 16]
Nephrectomy leaves the child with 1 functional kidney. In almost all patients, the remaining kidney can compensate and maintain adequate renal function. Additional treatment modalities after nephrectomy may damage several organs, such as the heart, lungs, liver, bones, and gonads. [17, 18] In addition, chemotherapy and radiation therapy can induce second malignant neoplasms. [19]
Renal complications
Children with Wilms tumor have a minimal risk for impaired renal function, primarily related to nephrectomy. In selected patients, (ie, those who receive radiation therapy), function of the remaining kidney can be further endangered. The development of compensatory postnephrectomy hypertrophy of the remaining kidney is well documented in patients with Wilms tumor.
NWTSG data suggest that most patients with unilateral Wilms tumor do not develop serious, long-term renal complications. By comparison, renal function can be impaired in those with bilateral disease. The most common cause of renal failure in patients with bilateral Wilms tumor is bilateral nephrectomy. Treatment-related injury (eg, radiation-induced damage, surgical complications) of the remaining kidney is the second leading cause of renal insufficiency.
In a study of 7950 patients in the National Wilms Tumor Studies (from 1969-2002), 100 patients, or approximately 1%, developed end-stage renal disease. The primary risk factors included stromal predominant histology, intralobar rests, age at diagnosis of younger than 24 months, metachronous bilateral Wilms tumor, and WT1 mutation etiology. [20] The particularly high frequency of renal failure associated with metachronous bilateral Wilms tumor was due to surgery for progressive Wilms tumor.
Hepatic complications
Several cytotoxic agents may damage the liver of patients treated for Wilms tumor, including dactinomycin and irradiation. Most early reports suggest that hepatic irradiation is the major etiologic factor in hepatic injury. However, reports have documented hepatic toxicity with the combination of vincristine and dactinomycin in nonirradiated children with Wilms tumor, suggesting that chemotherapeutic agents themselves can also damage the liver.
In the fourth NWTSG report, the incidence of hepatotoxicity was 2.8-14.3% in patients who did not receive irradiation. The fact that patients who received less dactinomycin than others (ie, those with relatively low-stage disease) had a low incidence of 2.8% suggests a dose-related toxicity for dactinomycin.
With the use of currently accepted radiotherapy techniques, radiation-induced hepatitis is rare in survivors of Wilms tumor.
Some patients with Wilms tumor have developed hepatic veno-occlusive disease (VOD). VOD is primarily a clinical diagnosis characterized by hepatomegaly or pain in the right upper quadrant, jaundice, ascites, and unexplained weight gain. The syndrome occurs in patients with Wilms tumor undergoing nephrectomy first and in those receiving combination chemotherapy before surgery, the standard approach that SIOP recommends. Although treatment for VOD is primarily supportive, the administration of chemotherapeutic agents can be resumed after the signs of VOD have disappeared.
Other complications of chemotherapy and radiation therapy
Congestive heart failure is a well-known complication of the administration of anthracyclines. Therefore, patients with Wilms tumor who receive anthracyclines, most commonly doxorubicin, should be monitored for cardiac dysfunction.
Because radiation therapy can affect pulmonary function, monitoring of pulmonary function is required in patients with metastatic Wilms tumors to the lung who are treated with bilateral pulmonary irradiation. The total lung capacity and vital capacity of patients receiving bilateral irradiation can be expected to decrease by 50-70% of the predicted values.
Women who received whole-abdomen irradiation in childhood can develop ovarian failure. Data clearly suggest that a high risk of adverse pregnancy outcomes should be considered in the counseling and prenatal care of women who received abdominal radiation therapy to treat a Wilms tumor. Male patients are at risk for testicular failure after whole-abdomen radiation therapy or certain types of chemotherapy, most notably that involving alkylating agents.
The effect of radiation therapy to the skeletal system is often predictable. Although radiation therapy may affect the growth of any given bone, the spine is most notably affected at doses of 20 Gy. A study from the University of Iowa showed a dose-response relationship in the induction of scoliosis and in the dose delivered. Most patients who received doses of more than 24 Gy with megavoltage beams developed asymptomatic scoliosis. Patients receiving current doses of 10-12 Gy may have a much reduced likelihood of developing scoliosis. [21]
Patients who survive Wilms tumor are at risk because inherited disposition and treatment (eg, chemotherapy, irradiation) can induce second malignant neoplasms. Most secondary malignant neoplasms reported (eg, bone tumors, breast and thyroid cancers) have occurred in irradiated areas. Nevertheless, certain chemotherapeutic agents, including doxorubicin, dactinomycin, and vincristine, may contribute to an increased risk for secondary malignancies.
Fifteen years after initial diagnosis, the cumulative incidence of a secondary malignant neoplasm in patients registered with the NWTSG was 1.6% and increasing. According to NWTSG investigators, abdominal irradiation increases the risk of a secondary malignant neoplasm and doxorubicin potentiates the radiation effect. Treatment for relapse further increased the risk for a secondary malignant neoplasm by a factor of 4-5.
In a study of 2,492 female subjects from the National Wilms Tumor Studies, Lange and colleagues found that female survivors of Wilms tumor had a 9.1-fold increased risk of developing invasive breast cancer and had an estimated cumulative risk of invasive breast cancer at age 40 of 4.5%. A total of 29 cases of invasive breast cancer were identified among 28 participants, all but four occurring before age 40. [22]
Relative to the general population, the risk was highest among women who had been treated with chest radiotherapy, who had a 27.6-fold increase and a cumulative risk at age 40 of 14.8%. Of the women who developed breast cancer after chest radiotherapy, most received doses of 12 Gy; the remainder received higher doses. No significant difference was found in breast cancer rates between women treated or not treated with abdominal irradiation. [22]
A report from the Childhood Cancer Survivor Study cohort that included 23,601 childhood cancer survivors, reported that the 15-year cumulative incidence of at least one severe to fatal chronic health condition was 11.9% for Wilms tumor survivors diagnosed between 1990 and 1999 and 17.6% for survivors diagnosed between 1970 and 1979. [23]
Patient Education
The parents and patient must know that long-term follow-up care is essential because of the late effects of treatment. Advise them that chemotherapy and irradiation, as well as inherited disposition, can induce second malignant neoplasms, such as bone tumors and breast and thyroid cancers.
Heart failure is a well-known complication of the administration of anthracyclines. Thus, patients with Wilms tumor who receive anthracyclines, most commonly doxorubicin, should be monitored for cardiac dysfunction.
Note that as many as one third of patients with Wilms tumor present with high blood pressure. Their blood pressure usually normalizes after nephrectomy, but they occasionally require prolonged therapy.
No precautions regarding activity are advised, although the patient and his or her parents should be aware that the patient will have only 1 kidney after therapy. Activities that carry an inherent risk of kidney injury, such as boxing and hockey, should be avoided.
-
CT scan in a patient with a right-sided Wilms tumor with favorable histology.
-
CT scan of child with a stage IV Wilms tumor with favorable histology. Note the bilateral pulmonary metastases.
-
Gross nephrectomy specimen shows a Wilms tumor pushing the normal renal parenchyma to the side.
Tables
Stage |
Relapse-Free Survival, % |
Overall Survival, % |
I |
92 |
98 |
II |
85 |
96 |
III |
90 |
95 |
IV |
80 |
90 |
Stage and Histology |
Surgery |
Chemotherapy |
Radiation Therapy* |
Stage I or II favorable histology without loss of heterozygosity (LOH) 1p and 16q† |
Nephrectomy |
Vincristine, dactinomycin |
No |
Stage I or II favorable histology with LOH 1p and 16q |
Nephrectomy |
Vincristine, dactinomycin, doxorubicin |
No |
Stage III and IV favorable histology without LOH 1p and 16q |
Nephrectomy |
Vincristine, dactinomycin, doxorubicin |
Yes |
Stage III and IV favorable histology with LOH 1p and 16q |
Nephrectomy |
Vincristine, dactinomycin, doxorubicin, cyclophosphamide, etoposide |
Yes |
* The current dose for radiation therapy for favorable histology Wilms tumor is approximately 1080 cGy for the abdomen and 1200 cGy for the lung. [29] Postoperative radiotherapy is started within 14 days of nephrectomy. [30] Patients with stage IV favorable histology Wilms tumor and lung metastases whose pulmonary lesions do not disappear after 6 weeks of chemotherapy receive whole-lung radiation therapy. † Some evidence suggests that certain children with stage I disease and favorable histology do well with nephrectomy alone. [31] Children younger than 24 months with small (< 550 g) Wilms tumors with favorable histology are noted in the current COG protocol. |
|||
Stage and Type of Wilms Tumor |
Imaging Studies |
Off-Treatment Schedule |
Stages I, II, and III with favorable histology; stages I, II, and III with anaplastic histology |
Chest radiography |
6 wk and 3 mo after surgery, then every 3 mo (5 times), then every 6 mo (3 times), then yearly (2 times) |
All stages in patients aged < 48 mo at diagnosis with nephrogenic rests |
Abdominal ultrasonography |
Every 3 mo for 6 y |
All stages in patients aged >48 mo at diagnosis with nephrogenic rests |
Abdominal ultrasonography |
Every 3 mo for 4 y |
Stages I and II with favorable histology |
Abdominal ultrasonography |
Yearly (6 times) |
Stage III with favorable histology |
Abdominal ultrasonography |
6 wk and 3 mo after surgery, then every 3 mo (5 times), then every 6 mo (3 times), then yearly (2 times) |
All stages with unfavorable histology |
Abdominal ultrasonography |
Every 3 mo (4 times), then every 6 mo (4 times) |
* Subsequent imaging studies should be performed as clinically indicated. |
||







