Overview
Waardenburg syndrome (WS) is named after the Dutch ophthalmologist Petrus Johannes Waardenburg, who, in 1947, first described a patient with hearing loss, dystopia canthorum (ie, lateral displacement of the inner canthi of the eyes), and retinal pigmentary differences. In 1951, after identifying other patients with similar symptoms, Waardenburg defined the syndrome now classified as WS type 1 (WS1). [1] Findings in WS1 include hearing loss, dystopia canthorum, and pigmentary abnormalities of the hair, skin, and eyes (see the images below).
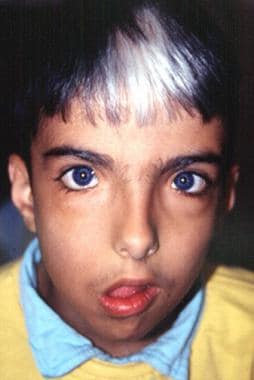 Marked facial asymmetry, lagophthalmos, drooping right corner of mouth. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
Marked facial asymmetry, lagophthalmos, drooping right corner of mouth. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
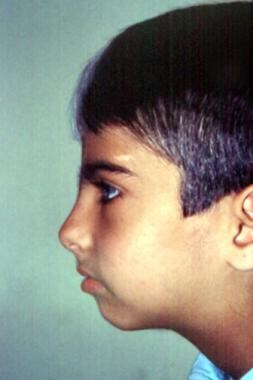 Visage in profile demonstrates absence of nasofrontal angle, eyebrow hypertrichosis, upturned nasal tip, and shortened upper lip with pronounced cupid's bow. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
Visage in profile demonstrates absence of nasofrontal angle, eyebrow hypertrichosis, upturned nasal tip, and shortened upper lip with pronounced cupid's bow. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
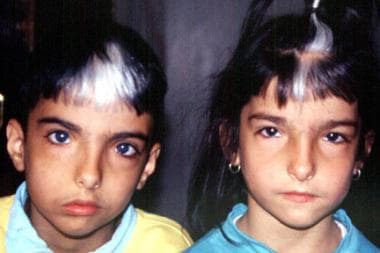 Brother and sister with Waardenburg syndrome. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
Brother and sister with Waardenburg syndrome. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
In 1971, Arias defined the phenotype of WS type 2 (WS2), which includes all of the WS1 features except dystopia canthorum. [2] Both WS1 and WS2 are transmitted as autosomal dominant conditions with interfamilial and intrafamilial variability.
Two far rarer variant forms of WS have also been identified. WS type 3 (WS3), or Klein-Waardenburg syndrome, includes features of WS in association with severe contractures. WS type 4 (WS4), or Waardenburg-Shah syndrome, has features of WS in association with Hirschsprung disease. WS4 is a heterogeneous disorder with either autosomal recessive or autosomal dominant inheritance.
WS, which is believed to account for 2-5% of patients with congenital hearing loss, has an estimated prevalence of 1 in 42,000 persons (or 0.24 per 10,000 persons). The actual prevalence, however, may be as high as 1.19-2.08 per 10,000 persons. [3]
Gene Mutations
The various forms of Waardenburg syndrome (WS), a neurocristopathy, arise from mutations in multiple genes. [4] The 6 genes involved in WS are PAX3 (encoding the paired box 3 transcription factor), MITF (microphthalmia-associated transcription factor), EDN3 (endothelin 3), EDNRB (endothelin receptor type B), SOX10 (encoding the Sry bOX10 transcription factor), and SNAI2 (snail homolog 2), with different frequencies. [5]
Evidence suggests that the MITF gene transactivates the tyrosinase gene, which is involved in melanocyte differentiation. PAX3 belongs to a family of paired-domain proteins that bind DNA and regulate gene expression; its molecular mechanism remains unclear. A study by Watanabe et al showed that PAX3 transactivates the MITF promoter. [6] Therefore, mutations in the PAX3 gene could affect regulation of the MITF gene, leading to abnormalities of melanocyte differentiation. A recent work identified 15 novel and 4 previously published heterozygous mutations in PAX3 and MITF. [7] Sequencing and copy number analysis of both PAX3 and MITF should be considered as part of the routine molecular diagnostic evaluation of these patients.
Waardenburg syndrome type 1
Most, if not all, cases of WS1 are caused by mutations in the PAX3 gene located on chromosome band 2q35. Deletions, frame shifts, splice site, and nonsense mutations, as well as whole gene deletions, have been reported. WS1 may be inherited in an autosomal dominant pattern or may be the result of a de novo mutation.
Two nonsense PAX3 mutations were identified in Chinese patients with WS1. One is heterozygous for a novel nonsense mutation, S209X, and the other is heterozygous for a previously reported mutation in the European population, R223X. [8] Both mutations created stop codons leading to truncation of the PAX3 protein. Novel mutations of PAX3, MITF, and SOX10 genes have been described in Chinese patients with WS1 or WS2. A novel PAX3 heterozygous mutation of c.372-373delGA (p.N125fs) was found that gave rise to a frameshift and truncation of the PAX3 protein. [9]
Waardenburg syndrome type 2
Mutations in the MITF gene, located on chromosome band 3p14.1-p12.3, cause some cases of WS2. Deletions, missense, splice site, and nonsense mutations have been reported. These mutations may be inherited in an autosomal dominant pattern or may be de novo. Other cases of WS2 have been linked to another locus on 1p21-p13.3, still others remain unlinked to either locus. Six novel variants were identified in 13 WS2 patients from six unrelated Chinese families by next-generation sequencing. Mutations in SOX10 and MITF were found to be the two major causes of associated deafness. [10]
Up to 20 mutations in the SOX10 gene have been linked to WS2. A novel dominant mutation in this gene was delineated in a Chinese family. [11]
Waardenburg syndrome type 3
Mutations in PAX3 have also been found in patients with a WS3 phenotype. WS3 may be inherited as a dominant disorder. In some cases, WS3 may be a manifestation of homozygous mutations of this gene.
Waardenburg syndrome type 4
WS4, which has been studied with a zebrafish model, [12] is caused by homozygous mutations in either the EDN3 gene or the EDNRB gene; heterozygous mutations in either of these genes cause isolated Hirschsprung disease. Sangkhathat et al reported that whereas homozygous mutations of EDNRB may result in WS4 and mutated heterozygotes manifest isolated Hirschsprung disease in lower penetrance, findings in a family were consistent with previous observations that the full spectrum of WS4 occurred to the mutated homozygotes. [13]
Heterozygous mutations in the SOX10 gene also reportedly cause WS4. [14] The SOX10 gene interacts with PAX3 in regulating the MITF gene. SOX10 mutations are associated with a more severe phenotype known as PCWH, consisting of peripheral demyelinating neuropathy, central dysmyelinating leukodystrophy, WS, and Hirschsprung disease. [15] Chronic constipation in WS may also represent a SOX10 mutation. [16] An association may exist between homozygous mutations in MITF and WS4. [17]
Associated abnormalities
Microdeletions or contiguous gene defects may be involved in the pathogenesis of other malformations associated with this syndrome. [18]
Waardenburg anophthalmia syndrome has also been described in children with a homozygous mutation in the SPARC -related modular calcium-binding protein 1 gene (SMOC1). [19]
Mutations in the genes PAX3 and MITF have been described in WS, which is clinically characterized by congenital hearing loss and pigmentation anomalies.
-
Marked facial asymmetry, lagophthalmos, drooping right corner of mouth. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
-
Visage in profile demonstrates absence of nasofrontal angle, eyebrow hypertrichosis, upturned nasal tip, and shortened upper lip with pronounced cupid's bow. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.
-
Brother and sister with Waardenburg syndrome. Image courtesy of Dourmishev LA et al, Cutis 1999; 63:139-40. Copyright 1999, Quadrant Healthcom, Inc.









