Practice Essentials
Although referred to as a single disease, von Willebrand disease (VWD) is in fact a family of bleeding disorders caused by an abnormality of the von Willebrand factor (VWF). von Willebrand disease is the most common hereditary bleeding disorder. [1, 2]
The condition was first described by Erik Adolf von Willebrand in 1926, who called it pseudohemophilia because of the fact that his first patient was a female. It took several decades before the actual nature of this bleeding disorder was understood. von Willebrand disease is a congenital bleeding disorder characterized by a lifelong tendency toward easy bruising, frequent epistaxis, and menorrhagia.
Signs of von Willebrand disease
The physical examination findings may be normal in von Willebrand disease, but the following may be present:
-
Increased bruises
-
Mucosal bleeding
Workup in von Willebrand disease
Screening tests for von Willebrand disease include the following:
-
Complete blood count (CBC)
-
Template bleeding time
-
Prothrombin time (PT)
-
Activated partial thromboplastin time (aPTT)
Specific assays include the following:
-
von Willebrand factor levels
-
Factor VIII (FVIII) activity
-
von Willebrand factor activity (ristocetin cofactor)
-
von Willebrand factor antigen
In multimer analysis to determine the physical structure of von Willebrand factor (ie, whether high–molecular-weight multimers are present), plasma is electrophoresed through agarose gel. The presence or absence of high–molecular-weight von Willebrand factor is used to classify von Willebrand disease.
Management of von Willebrand disease
Desmopressin (1-deamine-8-D-arginine vasopressin [DDAVP]) has become a mainstay of therapy for most patients with mild von Willebrand disease.
Pathophysiology
von Willebrand disease is due to an abnormality, either quantitative or qualitative, of the von Willebrand factor, which is a large multimeric glycoprotein required for normal platelet adhesion. It also functions as the carrier protein for factor VIII (FVIII). [3] As such, von Willebrand factor functions in both primary (involving platelet adhesion and platelet plug formation) and secondary (involving FVIII) hemostasis. In primary hemostasis, von Willebrand factor attaches to platelets by its specific receptor to glycoprotein Ib on the platelet surface and acts as an adhesive bridge between the platelets and damaged subendothelium at the site of vascular injury. In secondary hemostasis, von Willebrand factor protects FVIII from degradation and delivers it to the site of injury.
von Willebrand factor is composed of dimeric subunits that are linked by disulfide bonds to form complex multimers of low, intermediate, and high molecular weights. The small multimers function mainly as carriers for FVIII.
High–molecular weight multimers have higher numbers of platelet-binding sites and greater adhesive properties. Each multimeric subunit has binding sites for the receptor glycoprotein Ib on nonactivated platelets and the receptor glycoprotein IIb/IIIa on activated platelets. This facilitates both platelet adhesion and platelet aggregation, making high molecular weight multimers most important for normal platelet function. See the image below.
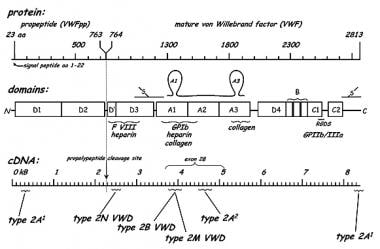 Structure and domains of von Willebrand factor.
Structure and domains of von Willebrand factor.
von Willebrand disease types
von Willebrand disease can be classified into 3 main types.
Type 1 von Willebrand disease
Type 1 von Willebrand disease, which accounts for 70-80% of cases, is characterized by a partial quantitative decrease of qualitatively normal von Willebrand factor and FVIII. An individual with type 1 von Willebrand disease generally has mild clinical symptoms, and this type is usually inherited as an autosomal dominant trait; however, penetrance may widely vary in a single family. In addition, clinical and laboratory findings may vary in the same patient on different occasions. Typically, a proportional reduction in von Willebrand factor activity, von Willebrand factor antigen, and FVIII is observed in type 1 von Willebrand disease. [4]
Type 2 von Willebrand disease
Type 2 disease accounts for 15-20% of von Willebrand disease cases. It is a variant of the disease with primarily qualitative defects of von Willebrand factor. Type 2 von Willebrand disease can be either autosomal dominant or autosomal recessive. [5] Of the four described type 2 von Willebrand disease subtypes (ie, 2A, 2B, 2M, 2N), type 2A is by far the most common.
Type 2A von Willebrand disease is inherited as an autosomal dominant trait and is characterized by normal-to-reduced plasma levels of factor VIIIc (FVIIIc) and von Willebrand factor. Analysis of von Willebrand factor multimers reveals a relative reduction in intermediate and high molecular weight multimer complexes. The multimeric abnormalities are commonly the result of in vivo proteolytic degradation of the von Willebrand factor. The ristocetin cofactor activity is greatly reduced, and the platelet von Willebrand factor reveals multimeric abnormalities similar to those found in plasma.
Type 2B von Willebrand disease is also an autosomal dominant trait. This type is characterized by a reduction in the proportion of high molecular weight von Willebrand factor multimers, whereas the proportion of low–molecular weight fragments are increased. Patients with type 2B von Willebrand disease have a hemostatic defect caused by a qualitatively abnormal von Willebrand factor and intermittent thrombocytopenia. This is a gain of function mutation in the von Willebrand factor causing spontaneous binding to platelets and rapid clearance of both platelets and high molecular weight von Willebrand factor multimers. The abnormal von Willebrand factor has an increased affinity for platelet glycoprotein Ib.
The platelet count may fall further during pregnancy, in association with surgical procedures, or after the administration of desmopressin acetate (DDAVP). Although some investigators found DDAVP to be clinically useful in persons with type 2B von Willebrand disease , studies directed at excluding the 2B variant should be completed before DDAVP is used. Measurements of FVIIIc and von Willebrand factor in plasma vary; however, studies involving the use of titered doses of ristocetin reveal that aggregation of normal platelets is enhanced and induced by unusually small amounts of the drug (low dose ristocetin induced platelet aggregation assay- LD- RIPA).
In patients with the rare type 2M von Willebrand disease, laboratory results are similar to those of certain patients with type 2A von Willebrand disease. Type 2M von Willebrand disease is characterized by a decreased platelet-directed function that is not due to a decrease of high–molecular weight multimers. Laboratory findings show decreased von Willebrand factor activity, although von Willebrand factor antigen, FVIII, and multimer analysis are found to be within reference range. This is caused by a defect in the von Willebrand factor gene that produces decreased or absent binding to platelet glycoprotein Ib and is autosomal dominant in inheritance. The ratio of von Willebrand factor ristocetin cofactor activity to the von Willebrand factor antigen is usually less than 0.5-0.7 (as in types 2A and 2B). Normal multimers distinguish type 2M from types 2A and 2B.
Type 2N von Willebrand disease is also rare and is characterized by a markedly decreased affinity of von Willebrand factor for FVIII because of a genetic mutation in the factor VIII binding region of von Willebrand factor; this results in FVIII levels being reduced to usually around 5-15% of the reference range, with a half life of about 2 hours only. Other von Willebrand factor laboratory parameters (ie, von Willebrand factor antigen, ristocetin cofactor activity) are usually normal. The FVIII-binding defect in these patients is inherited in an autosomal recessive manner. Male patients type 2N–like findings are frequently mistaken for having mild hemophilia A. For this reason, type 2N von Willebrand disease should be considered in patients with FVIII deficiency (mild FVIII deficiency) and a bleeding disorder that is not clearly transmitted as an X-linked disease, as well as in patients who respond incompletely to hemophilia A therapy. Unfortunately, the confirmatory test, which is a specific assay of FVIII binding to von Willebrand factor for type 2N von Willebrand disease, is not routinely available, likely resulting in an underestimation of the true frequency of this subtype. [6]
Type 3 von Willebrand disease
Type 3 von Willebrand disease is the rarest and most severe form of the condition. The disease is inherited as an autosomal recessive trait, and symptomatic patients are homozygous or combined heterozygous. Type 3 von Willebrand disease is characterized by marked deficiencies of both von Willebrand factor and FVIIIc (2-10%) in the plasma, the absence of von Willebrand factor from both platelets and endothelial cells, and a lack of response to DDAVP. Type 3 von Willebrand disease is also characterized by severe clinical bleeding, which is characteristic of both the primary hemostatic defects (superficial and mucous membrane bleeds) and the secondary hemostatic defects (joint and muscle bleeds) and is inherited as an autosomal recessive trait. Consanguinity is common in kindreds with this variant. Multimeric analysis of the small amount of von Willebrand factor present yields variable results, in some cases revealing only small multimers, if any.
Epidemiology
Frequency
United States
von Willebrand disease is estimated to affect about 1% of the population. [7]
International
Prevalence worldwide is estimated at 0.9-1.3%.
Mortality/Morbidity
The morbidity in individuals with von Willebrand disease varies. Many children with von Willebrand disease are asymptomatic. Some of these children have cutaneous and/or mucous membrane bleeding (eg, easy bruising, epistaxis).
Menorrhagia is a common symptom in females with von Willebrand disease. It occurs in more than 50% of women with von Willebrand disease and may be the only clinical manifestation of the disease.
The rare type 3 von Willebrand disease can manifest with severe bleeding symptoms similar to those of mild to moderate hemophilia A (eg, hemarthrosis, intramuscular bleeding).
A Swedish study, by Holm et al, found that compared with controls, patients with von Willebrand disease had a higher risk of hospitalization associated with cardiovascular disease (CVD; 1.3 fold) but a lower risk of CVD-linked mortality (0.4 fold). According to the investigators, this suggests that von Willebrand factor deficiency protects against arterial thrombosis even in the presence of atherosclerosis. [8]
Race
No influence of ethnicity on the prevalence of von Willebrand disease has been reported.
Sex
von Willebrand disease affects males and females in equal numbers.
Age
von Willebrand disease is a congenital bleeding disorder and can be diagnosed at any age.
-
Structure and domains of von Willebrand factor.