Background
In 1929, von Gierke provided the initial description of glycogen-storage disease type I (GSD I) from autopsy reports of 2 children whose large livers contained excessive glycogen. He also reported similar findings in the kidneys. Both children had frequent nosebleeds before their deaths, consistent with histories documented in current patients.
In 1952, Cori and Cori reported 6 similar patients. [1] Two of the patients had almost total deficiency of hepatic glucose-6-phosphatase, whereas the remaining 4 had normal enzyme activity. These authors recognized that defects in the enzymology of hepatic glycogen-storage disease may cause a heterogeneous group of disorders. However, the mystery of patients with these clinical symptoms (despite normal phosphatase activity) remained unsolved until 1978, when Narisawa et al identified a defect in intracellular transport of the enzyme substrate. [2]
In recognition of the original clinical description of the disease, the type I Cori classification has been preserved; glycogen-storage disease type Ia (GSD Ia) designates the true enzyme defect, and glycogen-storage disease type Ib (GSD Ib) designates the intracellular transport defect. Because free glucose is the product of the hepatic glucose-6-phosphatase reaction, either type leads to accumulation of liver glycogen, accompanied by fasting hypoglycemia. Hepatomegaly, the natural consequence of glycogen accumulation, is the clinical hallmark of the disease.
In 2014, the American College of Genetics and Genomics issued an updated practice guideline on the diagnosis and management of types Ia and Ib. [3]
More than 80 discrete mutations have been identified for both types Ia and Ib.
Pathophysiology
The liver loses its capacity as a glucose-homeostatic organ because of a fundamental inability to release free glucose. Glycogen-storage disease type I (subtypes Ia and Ib) is one of the few genetic-biochemical causes of hypoglycemia in newborns. The usual homeostatic mechanism cannot halt the rapid drop in blood glucose levels that normally occurs during the first several hours after birth (reflecting consumption of maternal glucose), and the decrease continues. This decrease in circulating glucose can be precipitous, resulting in no measurable blood level. Seizures, cyanosis, and apnea may ensue. In older children, repeated episodes of hypoglycemia may result in brain damage, as measured on performance testing and assessment of brainstem auditory-evoked potentials.
In the hepatocyte, the glycogen catabolic machinery normally responds to stimuli caused by hypoglycemia (eg, neural, hormonal), ending in a flood of glucose-6-phosphate that cannot be released from the cell. However, glucose-6-phosphate is also the substrate for glycolysis and produces lactate. Lactate exits the hepatocyte, causing clinically significant lactic acidemia in proportion to the degree of stimulus for glycogen breakdown. The accumulation of lactic acid in blood can cause true acidosis with a large anion gap, a characteristic of glycogen-storage disease type I.
The immense increase in the intracellular phosphorylated intermediate compounds of glycolysis concurrently inhibits rephosphorylation of adenine nucleotides, activating the nucleic acid degradation pathway and resulting in increased uric acid, the end product. Hyperuricemia can reach levels that require use of xanthine oxidase inhibitors to prevent nephrolithiasis. Nephrolithiasis secondary to increased uric acid is a constant threat to patients with poorly controlled disease.
Severe hypoglycemia stimulates epinephrine secretion, which activates lipoprotein lipase and the release of free fatty acids. These fatty acids are transported to the liver, where they are used for triglyceride synthesis and are exported as very-low-density lipoprotein (VLDL), which is elevated in these patients. Paradoxically, even in the face of hypoglycemia, patients with glycogen-storage disease I do not develop significant ketosis because the abundance of acetyl coenzyme A (CoA) derived from glycolysis activates the acetyl CoA carboxylase enzyme that produces malonyl CoA in the first step of fatty acid synthesis. Because malonyl CoA inhibits transport of fatty acid into the mitochondrion, beta-oxidation of fatty acids for energy to support the hypoglycemic cells does not occur. This causes a continuing drop in blood glucose levels and explains the absence of ketone bodies.
The lipid abnormalities, which include hypercholesterolemia (decreased high-density lipoprotein [HDL] cholesterol and increased low-density lipoprotein [LDL] cholesterol), together with the characteristic hypertriglyceridemia do not cause premature atherosclerotic lesions in affected individuals. Recent evidence suggests that sera from patients with glycogen-storage disease Ia are able to more efficiently promote scavenger receptor class B type I–mediated cellular cholesterol efflux and that an increased antioxidant effect of these sera is directly related to the increased urate concentration. [4]
Notwithstanding these data, the results of a 2015 prospective multisite clinical study suggest a statistical correlation between progression of renal disease and serum lipid levels over time. [5] In addition, a 2009 report suggests that affected individuals may sustain an increase in carotid artery intimal thickness and arterial dysfunction. [6]
Nosebleeds experienced by the patients in von Gierke's report probably resulted from the bleeding tendency characteristic of glycogen-storage disease Ia and Ib. This tendency resembles von Willebrand disease and suggests alterations in membrane glycoprotein synthesis. Although such changes have been found, no definitive explanation addresses how these alterations actually cause defective platelet aggregation.
Patients with glycogen-storage disease Ib are susceptible to gram-positive infections. Neutrophils from patients with glycogen-storage disease Ib have a significantly impaired respiratory-burst response to stimuli compared with type Ia neutrophils. Because the respiratory burst generates superoxide, which is a major defense against gram-positive organisms, a defect in this response would be expected to render the neutrophils susceptible, causing neutropenia and diminishing the individual's resistance to infection. Evidence suggests that microsomal transport of glucose-6-phosphate has a role in antioxidant protection of the neutrophil; therefore, a genetic defect in the transporter could impair cellular function and lead to apoptosis, as shown in the image below.
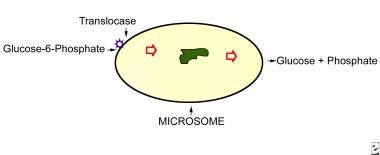 Microsome is shown in relation to the substrate, glucose-6-phosphate, which has been released from cytosolic glycogen. This substrate is transferred across the microsomal membrane by the protein translocase, where by glucose-6-phosphatase acts on it to release free glucose and inorganic phosphate. Patients with glycogen-storage disease type Ia are genetically deficient in glucose-6-phosphate activity, while those affected with glycogen-storage disease type Ib lack translocase.
Microsome is shown in relation to the substrate, glucose-6-phosphate, which has been released from cytosolic glycogen. This substrate is transferred across the microsomal membrane by the protein translocase, where by glucose-6-phosphatase acts on it to release free glucose and inorganic phosphate. Patients with glycogen-storage disease type Ia are genetically deficient in glucose-6-phosphate activity, while those affected with glycogen-storage disease type Ib lack translocase.
Growth is generally impaired in patients with glycogen-storage disease I, although growth can be improved with good dietary therapy in most patients. However, growth remains unimproved by treatment in some patients; the endocrine parameters of growth in this group are not measurably different from the larger number of patients.
Several studies have also documented a decreased bone mineral density in some patients with glycogen-storage disease I. One such investigation reported no correlation between bone mineral density and turnover markers, indicating an uncoupling of bone turnover in patients with glycogen-storage disease. [7, 8]
The cause of severe anemia in the absence of renal function compromise in children with glycogen-storage disease I has remained unclear. Some have recently proposed that hepcidin production by hepatic adenomas plays a central role in patients with glycogen-storage disease I. Hepcidin is a peptide hormone that is also a key regulator of the egress of cellular iron; in excess, it may interfere with intestinal iron transport as well as iron release from macrophages.
Although extremely rare, glycogen-storage disease subtypes Ic and Id have occurred. Individuals with these forms probably have unusual mutations in the translocase gene (11q23).
Epidemiology
Frequency
United States
The lack of newborn screening precludes reliable estimates of the incidence. The only estimates are for glycogen-storage diseases as a group, which suggest an incidence of 1 case per 20,000-25,000 births. Glycogen-storage disease I is unlikely to occur more frequently than 1 case in 50,000 infants.
In an odd quirk, clinical symptoms of biotinidase deficiency in patients with underlying glycogen-storage disease Ia have been associated with marked elevations in biotinidase levels on serum assay. However, mass screening for biotinidase deficiency does not reveal elevated activity.
Mortality/Morbidity
Affected newborns are at risk for all neonatal hypoglycemic complications of glycogen-storage disease type I. (Older children under treatment may experience symptoms identical to those listed below with impending hypoglycemia.)
Hypoglycemic complications are entirely nonspecific and include the following:
-
Twitching
-
Cyanosis
-
Seizures
-
Irritability
-
Apathy
-
Hypotonia
-
Hypothermia
-
Apnea
-
Coma (may be secondary to cerebral edema from combined hypoglycemia and hypoxia secondary to hypoglycemic seizures)
Long-term consequences of glycogen storage include the following:
-
Hepatic adenomas
-
Hepatocellular carcinoma
-
Progressive renal insufficiency
-
Hyperuricemic nephrocalcinosis
-
Hyperlipidemic xanthomas
-
Hypoglycemic brain damage
In glycogen-storage disease Ib, secondary consequences of neutrophilic abnormalities include multiple and recurrent infections, brain abscess, and pseudocolitis.
Sex
Glycogen-storage disease Ia and Ib occur with equal frequency in both sexes.
Age
As genetic disorders, both types are present at conception, with clinical onset at birth.
-
Microsome is shown in relation to the substrate, glucose-6-phosphate, which has been released from cytosolic glycogen. This substrate is transferred across the microsomal membrane by the protein translocase, where by glucose-6-phosphatase acts on it to release free glucose and inorganic phosphate. Patients with glycogen-storage disease type Ia are genetically deficient in glucose-6-phosphate activity, while those affected with glycogen-storage disease type Ib lack translocase.









