Background
A ventricular septal defect (VSD) is a hole or a defect in the septum that divides the two lower chambers of the heart, resulting in communication between the ventricular cavities. A VSD may occur as a primary anomaly, with or without additional major associated cardiac defects. It may also occur as a single component of a wide variety of intracardiac anomalies, including tetralogy of Fallot (TOF), complete atrioventricular (AV) canal defects, transposition of great arteries, and corrected transpositions.
In this article, the term ventricular septal defect refers to an isolated VSD, or a defect in a heart with AV concordance. That is, the atria are attached to the correct ventricle and the normally related arteries (great arteries arising from the appropriate ventricle [ie, an otherwise normal heart]), with no other major lesions. An isolated VSD occurs in approximately 2-6 of every 1000 live births and accounts for more than 20% of all congenital heart diseases. After bicuspid aortic valves, VSDs are the most commonly encountered congenital heart defects.
VSDs were first clinically described by Roger in 1879 [1] ; the term maladie de Roger is still used to refer to a small asymptomatic VSD. In 1898, Eisenmenger described a patient with VSD, cyanosis, and pulmonary hypertension. This combination has been termed the Eisenmenger complex. Pulmonary vascular disease and cyanosis in combination with any other systemic-to-pulmonary connection has been called the Eisenmenger syndrome. [2]
Heath and Edwards described the morphologic changes associated with pulmonary vascular disease in 1958, and their 6 categories of vascular change have remained the standard of comparison up to the present day. [3]
The symptoms and physical findings associated with ventricular septal defects (VSDs) depend on the size of the defect and the magnitude of the left-to-right shunt, which, in turn, depends on the relative resistances of the systemic and pulmonary circulations (see Presentation).
Chest radiography, magnetic resonance imaging (MRI), and electrocardiography (ECG) may all provide useful information in the workup of a VSD. Although cardiac catheterization was a standard part of the evaluation in the past, detailed echocardiography is now the diagnostic imaging modality of choice (see Workup).
Children with small VSDs are asymptomatic and have an excellent long-term prognosis. Neither medical therapy nor surgical therapy is indicated. In children with moderate or large VSDs, medical therapy is indicated to manage symptomatic congestive heart failure (CHF) because some VSDs may become smaller with time, although uncontrolled CHF symptoms with growth failure is an indication for surgical repair. Neither the age nor the size of the patient is prohibitive in considering surgery (see Treatment).
Anatomy
A ventricular septal defect (VSD) is a developmental defect of the interventricular septum, wherein communication between the cavities of the two ventricles is observed. Since 1979, real-time two-dimensional (2D) echocardiography has dramatically improved the noninvasive anatomic assessment of ventricular septal defect.
Embryology
At 4-8 weeks’ gestation, the single ventricular chamber is effectively divided into two chambers. This division is accomplished with the fusion of the membranous portion of the interventricular septum, the endocardial cushions, and the bulbous cordis (the proximal portion of the truncus arteriosus).
The muscular portion of the interventricular septum grows cephalad as each ventricular chamber enlarges, eventually meeting with the right and left ridges of the bulbous cordis. The right ridge fuses with the tricuspid valve and the endocardial cushions, separating the pulmonary valve from the tricuspid valve. The left ridge fuses with a ridge of the interventricular septum, leaving the aortic ring in continuity with the mitral ring.
The endocardial cushions develop concomitantly and finally fuse with the bulbar ridges and the muscular portion of the septum. The fibrous tissue of the membranous portion of the interventricular septum makes the final closure and separates the two ventricles.
Structure of interventricular septum
The interventricular septum is a curvilinear complex structure that can be divided into four zones on the basis of anatomic landmarks in the right ventricle (RV).
The RV has many heavy trabeculations. The stoutest of these is a Y-shaped bundle known as the trabecula septomarginalis, which proceeds toward the apex and which gives rise to the moderator band that courses transversely near the apex. The trabecula septomarginalis is an important structure that helps in identifying the RV, regardless of its location in the chest. The two limbs of the Y travel superiorly, with the anterior (parietal) limb supporting the pulmonic valve and the posterior limb (septal band) extending to the membranous septum.
The four parts of the interventricular septum are as follows (see the image below) [4] :
-
Inlet septum: This region is smooth-walled and extends from the septal attachments of the tricuspid valve to the distal attachments of the tricuspid tensor apparatus; it is also called the AV canal septum.
-
Trabecular septum: This apical trabecular zone separates the coarse trabeculations of the RV from the fine ones seen in the left ventricle (LV); it is also known as the muscular septum or the ventricular sinus septum.
-
Outlet (infundibular) septum: This smooth-walled region is separated from the trabeculated portion of the RV by the septal band of the trabecula marginalis; it is also referred to as the parietal band or the distal conal septum, and defects in this area may be termed conal septal defects.
-
Membranous septum: This region, the last and the smallest part of the interventricular septum, lies between the anterior and the septal tricuspid leaflets and below the right and the noncoronary cusps of the aortic valve.
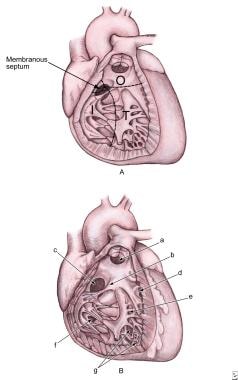 Ventricular Septal Defects. A: Image shows a ventricular septum viewed from the right side. It has the following four components: inlet septum from the tricuspid annulus to the attachments of the tricuspid valve (I); trabecular septum from inlet to apex and up to the smooth-walled outlet (T); outlet septum, which extends to the pulmonary valve (O); and membranous septum. B: Anatomic positions of the defects are as follows: outlet defect (a); papillary muscle of the conus (b); perimembranous defect (c); marginal muscular defects (d); central muscular defects (e); inlet defect (f); and apical muscular defects (g).
Ventricular Septal Defects. A: Image shows a ventricular septum viewed from the right side. It has the following four components: inlet septum from the tricuspid annulus to the attachments of the tricuspid valve (I); trabecular septum from inlet to apex and up to the smooth-walled outlet (T); outlet septum, which extends to the pulmonary valve (O); and membranous septum. B: Anatomic positions of the defects are as follows: outlet defect (a); papillary muscle of the conus (b); perimembranous defect (c); marginal muscular defects (d); central muscular defects (e); inlet defect (f); and apical muscular defects (g).

The three muscular components of the interventricular septum described above abut on the membranous septum and fan out from it as triangles, with the apices touching this septum.
In the normal heart, the tricuspid and mitral valves are attached to the ventricular septum at different levels, so that the tricuspid-valve attachment is apically displaced relative to the mitral-valve attachment. Therefore, a portion of the interventricular septum, called the AV septum, lies between the right atrium (RA) and the LV. This portion consists of a membranous part anteriorly and a muscular part posteriorly and is usually present in most hearts with an isolated VSD.
In the anterior aspect, the tricuspid-valve attachment divides the area of membranous septum into an interventricular component (between the LV and the RV) and an AV component (between the LV and the RA). When a VSD is isolated, the AV component of membranous septum is usually intact.
Classifications of ventricular septal defects
Many classifications of VSDs have been proposed. The following is a summary of an underlying classification that is surgically and clinically useful (see the image below).
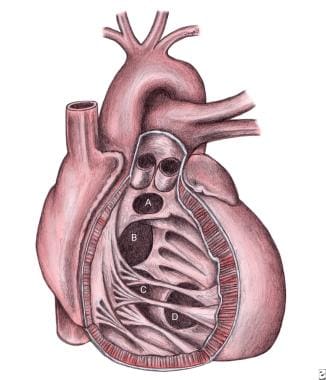 Ventricular Septal Defects. Schematic representation of the location of various types of ventricular septal defects (VSDs) from the right ventricular aspect. A = Doubly committed subarterial ventricular septal defect; B = Perimembranous ventricular septal defect; C = Inlet or atrioventricular canal-type ventricular septal defect; D = Muscular ventricular septal defect.
Ventricular Septal Defects. Schematic representation of the location of various types of ventricular septal defects (VSDs) from the right ventricular aspect. A = Doubly committed subarterial ventricular septal defect; B = Perimembranous ventricular septal defect; C = Inlet or atrioventricular canal-type ventricular septal defect; D = Muscular ventricular septal defect.
Perimembranous (infracristal, conoventricular) VSDs lie in the LV outflow tract just below the aortic valve. Because they occur in the membranous septum with defects in the adjacent muscular portion of the septum, they are subclassified as perimembranous inlet, perimembranous outlet, or perimembranous muscular. These are the most common types of VSD and account for 80% of such defects.
Perimembranous VSDs are associated with pouches or aneurysms of the septal leaflet of the tricuspid valve, which can partially or completely close the defect. In addition, an LV-to-RA shunt may be associated with this defect.
Supracristal (conal septal, infundibular, subpulmonic, subarterial, subarterial doubly committed, outlet) VSDs account for 5-8% of isolated VSDs in the United States but 30% of such defects in Japan. These defects lie beneath the pulmonic valve and communicate with the RV outflow tract above the supraventricular crest and are associated with aortic regurgitation secondary to the prolapse of the right aortic cusp.
Muscular VSDs (trabecular) are entirely bounded by the muscular septum and are often multiple. The term Swiss-cheese septum has been used to describe multiple muscular VSDs. Other subclassifications depend on the location and include central muscular or midmuscular, apical, and marginal (when the defect is along the RV-septal junction). These VSDs account for 5-20% of all defects. Any single defect observed from the LV aspect may have several openings on the RV aspect.
Posterior (canal-type, endocardial cushion–type, AV septum–type, inlet, juxtatricuspid) VSDs lie posterior to the septal leaflet of the tricuspid valve. Although the locations of posterior VSDs are similar to those of VSDs observed with AV septal defects, they are not associated with defects of the AV valves. About 8-10% of VSDs are of this type.
Other anatomic considerations
The relation of the AV conduction pathways to the defect is important for surgical repair. The AV node occupies the apex of the triangle of Koch, which is limited posteriorly by the tendon of Todaro (a fibrous extension from the Eustachian valve), inferiorly by the os of the coronary sinus, and superiorly by the tricuspid valve annulus. The bundle of His arises from the AV node.
In perimembranous defects, the bundle of His lies in a subendocardial position as it courses along the posterior-inferior margin of the defect. In inlet defects, the bundle of His passes anterosuperiorly to the defect. In muscular VSDs and outlet defects, the risk of heart block is minimal because the bundle is remote from the defect.
Patients with subpulmonary conal defects usually have deficiency of muscular or fibrous support below the aortic valve with subsequent herniation of the right aortic leaflet. However, in patients with perimembranous VSDs and aortic insufficiency, it may be the right or the noncoronary cusp that prolapses.
Pathophysiology
A defect in the interventricular septum allows communication between the systemic and pulmonary circulations. As a result, flow moves from a region of high pressure to a region of low pressure—that is, from the LV to the RV (a left-to-right shunt). The pathophysiologic effects of a VSD derive from the hemodynamic effects secondary to a left-to-right shunt and from changes in the pulmonary vasculature.
Left-to-right shunt
A left-to-right shunt at the ventricular level has three hemodynamic consequences:
-
Increased LV volume load
-
Excessive pulmonary blood flow and elevated pulmonary artery pressures
-
Reduced systemic cardiac output
Blood flow through the defect from the LV to the RV results in oxygenated blood entering the pulmonary artery (PA). The addition of this extra blood to the normal pulmonary flow from the vena cava increases blood flow to the lungs and subsequently increases pulmonary venous return into the left atrium (LA) and ultimately into the LV. This increased LV volume results in LV dilatation and then hypertrophy. It increases end-diastolic pressure and consequently LA pressure, then raises pulmonary venous pressure.
The increased pulmonary blood flow raises pulmonary capillary pressure, which can increase pulmonary interstitial fluid. When this condition is severe, patients can present with pulmonary edema. Therefore, both PA pressure and pulmonary venous pressure are elevated in a VSD. The increase in pulmonary venous pressure is not seen with an atrial septal defect: LA pressures are low because as blood can readily exit from this chamber through the atrial communication.
Finally, as blood is shunted through the VSD away from the aorta, cardiac output decreases, and compensatory mechanisms are stimulated to maintain adequate organ perfusion. These mechanisms include increased catecholamine secretion and salt and water retention by means of the renin-angiotensin system.
The degree of the left-to-right shunt determines the magnitude of the changes described above. The left-to-right shunt depends on two factors, of which one is anatomic and the other physiologic.
The anatomic factor is the size of the VSD. (The location of the VSD is irrelevant in terms of the degree of the shunt.) In a normal heart, RV pressure is about 25-30% that of the LV. In a large VSD, this pressure difference is no longer maintained, because a large hole offers no resistance to blood flow. Consequently, these defects are called nonrestrictive VSDs.
However, in a small VSD, the normal pressure difference between the ventricles is maintained. These defects are called restrictive VSDs because blood flow across the defects is restricted, so that the normal pressure difference is maintained.
The physiologic factor is the resistance of the pulmonary vascular bed.
Changes in pulmonary vasculature
The terms pulmonary hypertension, high pulmonary resistance, and pulmonary vascular disease are often confused. Pulmonary hypertension merely indicates a high blood pressure in the pulmonary circuit; depending on the duration, it may be reversible. Pulmonary resistance is a function of numerous factors, including age, altitude, hematocrit, and diameter of the pulmonary arterioles.
A neonate has increased resistance secondary to the increase in the media of the pulmonary arterioles; this decreases the effective diameter of the vessels. In addition, neonates have a relative polycythemia. The elevated pulmonary resistance usually declines to adult levels by 6-8 weeks.
Pulmonary vascular disease is ultimately an irreversible condition and may occur over time in individuals with a large left-to-right shunt. It may also occur in the absence of a shunt; this condition is called primary pulmonary hypertension. A characteristic series of histologic changes ranging from grade I to grade VI has been described. [3] The ultimate consequences of pulmonary vascular obstructive disease are irreversible vascular changes and pulmonary resistance equal to or exceeding systemic resistance.
Natural history
The natural history of VSD has a wide spectrum and is directly proportional to the size, location, and restriction of the defect, ranging from spontaneous closure to congestive heart failure (CHF) or the development of pulmonary vascular disease without heart failure symptoms.
Spontaneous closure frequently occurs in children, usually by age 2 years. Closure is uncommon after age 4 years. Closure is most frequently observed in muscular defects (80%), followed by perimembranous defects (35-40%). Outlet VSDs have a low incidence of spontaneous closure, and inlet VSDs do not close.
Closure may occur by means of hypertrophy of the septum, formation of fibrous tissue, subaortic tags, apposition of the septal leaflet of the tricuspid valve, or (in rare cases) prolapse of a leaflet of the aortic valve. When perimembranous VSDs close because of development of fibrous tissue or the apposition of the tricuspid valve, an "aneurysm" of the interventricular septum may appear that is merely the redundant tricuspid valve leaflet.
A small VSD that does not spontaneously close is associated with an excellent prognosis. Patients are theoretically at risk for infective endocarditis, but small muscular VSDs pose no other adverse possibilities.
Small perimembranous VSDs, however, are associated with an increased risk of prolapse of the aortic cusp over time. In addition, a small but definite risk of malignant ventricular arrhythmia was reported in the Second Natural History Study. [5] This study group consisted of about 1000 patients (about 76% of the original cohort). The original cohort was the First Natural History Study, which included 1280 patients (mostly children) with VSDs admitted after cardiac catheterization between 1958 and 1969.
Wu et al reported a 45% incidence of LV-to-RA shunts and a 6% incidence of subaortic ridges during a 20-year follow-up of about 900 patients with perimembranous VSDs. [6] This group later reported an increased incidence of infective endocarditis in patients who had LV-to-RA shunts. [7]
Etiology
For the purposes of etiologic analysis, clustering the defect types mentioned earlier (see Anatomy) according to potential pathogenic mechanisms is beneficial. The following pathologic classification allows comparison of similar defects:
-
Subarterial VSDs can be classified as abnormalities of ectomesenchymal tissue migration
-
Perimembranous VSDs can be classified as abnormalities of intracardiac blood flow
-
Muscular VSDs can be classified as abnormalities in cell death
-
Type III inflow VSDs can be classified as abnormalities of the extracellular matrix and defects in the endocardial cushion
At present, a multifactorial etiology based on an interaction between hereditary predisposition and environmental influences is assumed to cause the defects. The following questions have relevance to children, their family, and their parents alike:
-
What caused a child’s heart defect?
-
What is the risk that other children and grandchildren in the family will have a heart defect?
Maternal factors
Maternal diabetes has long been recognized as a risk factor for congenital cardiovascular malformations (CCVMs). The risk of CCVMs remains high for infants of women with poorly controlled elevated phenylalanine levels.
No population-based data are available to define the range of risk alcohol consumption poses to the developing cardiovascular system. Investigators from the Baltimore-Washington Infant Study (BWIS) reported that maternal alcohol consumption was associated with only muscular ventricular septal defect. [8]
Genetic risk factors
The single largest determinant in the BWIS data set is the presence of a genetic risk factor defined as a previous occurrence of a congenital cardiovascular defect in the family. A family history of a cardiac or noncardiac defect in either a parent or a preceding sibling is a major risk factor.
The incidence of VSD in siblings of patients with the same malformation is about three times that of the general population. VSDs have been reported in identical twins, but the frequency of discordance is high, even in identical twins.
Familial congenital heart defects are often concordant by phenotype and developmental mechanism. Among cases with VSDs, previous occurrence of transposition, tetralogy of Fallot (TOF), and truncus arteriosus is higher than expected.
Genotype-phenotype correlation
The challenge for the next generation of pediatric cardiologists is to collaborate with geneticists to define genotype-phenotype correlations.
Regarding genetic counseling and prospects for prevention, the single greatest change in counseling regarding the recurrence risk for CCVMs is the recognition of familial and chromosomally based defects see the Table below). Thorough evaluation includes the following:
-
An accurate clinical diagnosis of the cardiovascular defect(s) organized in a hierarchy (this is necessary to specify the type of VSD)
-
Carefully detailed noncardiac defects
-
Careful family history of first-degree and second-degree relatives, including detailed analysis of pregnancy loss, racial origin, and consanguinity
-
A search for risk factors, such as gestational diabetes mellitus
Table. Aneuploid Syndromes Associated with Ventricular Septal Defect (Open Table in a new window)
Syndrome |
CCVM (%) |
Type of CCVM |
Del 4q, 21, 32 |
60 |
Ventricular septal defect, atrial septal defect |
Del 5p |
30-60 |
Ventricular septal defect |
Trisomy 13 |
80 |
Atrial septal defect, ventricular septal defect, TOF |
Trisomy 18, Edwards syndrome |
100 |
Ventricular septal defect, TOF, double-outlet right ventricle (DORV) |
Trisomy 21, Down syndrome |
40-50 |
Ventricular septal defect, atrioventricular canal (AVC) |
Del 22q11, DiGeorge syndrome (single gene etiology, autosomal dominant) |
50 |
Truncus arteriosus, TOF, ventricular septal defect |
Epidemiology
United States statistics
VSDs affect 2-7% of live births. The patient’s area of residence may influence the prevalence of known VSDs. For example, small muscular VSDs are most likely to be identified in urban locations, possibly because of ready access to sophisticated healthcare in these locations.
An echocardiographic study revealed a high incidence of 5-50 VSDs per 1000 newborns. The defects in this study were small restrictive muscular VSDs, which typically spontaneously close in the first year of life. [9]
VSDs are the most common lesion in many chromosomal syndromes, including trisomy 13, trisomy 18, trisomy 21, and relatively rare syndromes. However, in more than 95% of patients with VSDs, the defects are not associated with a chromosomal abnormality.
Sex- and race-related demographics
VSDs are slightly more common in female patients than in male patients (56% vs 44%). The incidence of abnormalities of ectomesenchymal tissue migration (ie, subarterial outlet VSD) is highest in boys.
Reports are inconclusive regarding racial differences in the distribution of VSDs. However, the doubly committed or outlet defect occurs is most common in the Asian population. These constitute 5% of the defects in the Unites States but 30% of those reported in Japan.
Prognosis
Children with small VSDs are asymptomatic and have excellent long-term prognoses. The outcome of medical therapy for children with moderate or large VSDs varies, as follows.
Many infants improve, showing evidence of a gradual decrease in the magnitude of the left-to-right shunt between the ages of 6 and 24 months. It is important to assess the cause of the decrease in left-to-right flow, which can reflect an increase in pulmonary vascular resistance (PVR), a decrease in the relative size of the defect, or the development of RV outflow tract hypertrophy, resulting in functional or anatomic obstruction.
Most children with VSDs remain in stable condition or improve after infancy. Heart failure rarely occurs after infancy. Anemia, respiratory infection, endocarditis, or the development of an associated lesion (eg, aortic insufficiency) can trigger a recurrence of symptoms.
A few patients who develop severe pulmonary vascular obstructive disease with predominant right-to-left shunts (Eisenmenger syndrome) at the time of referral require symptomatic therapy. Cyanosis progressively increases, and exercise capacity decreases. Select patients with large VSDs may be candidates for lung or heart-lung transplantation.
Red blood cell (RBC) reduction by means of partial-exchange transfusion may relieve symptoms associated with extreme polycythemia (eg, headache, extreme fatigue).
The surgical mortality is less than 1% for isolated VSDs.
Complications
Eisenmenger complex is the most severe complication of a large VSD. Fixed and irreversible pulmonary hypertension develops, resulting in reversal of the left-to-right shunt to a right-to-left shunt.
Secondary aortic insufficiency is associated with prolapse of aortic valve leaflets. It is rare in children younger than 2 years. This complication is observed only in 5% of patients with VSD. The incidence is higher in supracristal VSDs than in perimembranous VSDs.
The development of aortic regurgitation in association with doubly committed subarterial VSD is a well-known phenomenon. Aortic regurgitation is due to a poorly supported right coronary cusp combined with the Venturi effect produced by the VSD jet, resulting in cusp prolapse.
Right ventricular (RV) outflow tract obstruction was noted in 7% of a large cohort of VSD in France. [10] The investigators noted the obstruction to be infundibular. A later angiocardiographic study showed that the obstruction was most often secondary to anomalous muscle bundles and only rarely infundibular. [11]
Discrete fibrous subaortic stenosis is occasionally associated with a VSD. This complication is most often reported with perimembranous VSDs and can first appear after either spontaneous or surgical closure. Zielinsky et al concluded that anterior or posterior malalignment of the outlet or the conal septum is present in all patients with a VSD who develop discrete subaortic stenosis. [12]
Infective endocarditis is rare in children younger than 2 years. In the presence of infective endocarditis in the pulmonary circulation, it is important to record the patient’s history meticulously and to investigate the left-to-right shunt by means of echocardiography. With VSDs, both the systemic and pulmonary circulation may be affected; hence, vegetation manifests on both sides.
Embolization is expected despite the morphology of the vegetation. In general, vegetation more than 10 mm, particularly if pedunculated, should be regarded as an indication for surgical intervention, even in the absence of symptoms.
Infection is usually located at the ridge of the VSD itself or on the tricuspid or pulmonary valve leaflets.
Intramural VSD, in which interventricular communications occur through right ventricular free wall trabeculations, may occur following biventricular repair of conotruncal anomalies. [13] These types of VSDs have an associated postoperative morbidity, mortality, and longer postoperative hospital stays. [13]
-
Ventricular Septal Defects. A: Image shows a ventricular septum viewed from the right side. It has the following four components: inlet septum from the tricuspid annulus to the attachments of the tricuspid valve (I); trabecular septum from inlet to apex and up to the smooth-walled outlet (T); outlet septum, which extends to the pulmonary valve (O); and membranous septum. B: Anatomic positions of the defects are as follows: outlet defect (a); papillary muscle of the conus (b); perimembranous defect (c); marginal muscular defects (d); central muscular defects (e); inlet defect (f); and apical muscular defects (g).
-
Ventricular Septal Defects. Schematic representation of the location of various types of ventricular septal defects (VSDs) from the right ventricular aspect. A = Doubly committed subarterial ventricular septal defect; B = Perimembranous ventricular septal defect; C = Inlet or atrioventricular canal-type ventricular septal defect; D = Muscular ventricular septal defect.
-
Ventricular Septal Defects. Supracristal ventricular septal defect (VSD) on computed tomography scanning. Top image: Parasternal long-axis view shows the defect just below the aortic root. Middle image: The plane of sound is tilted to view the right ventricular (RV) outflow tract, and the defect is observed below the pulmonic valve. Bottom image: Parasternal short-axis view shows the ventricular septal defect between the aortic root (Ao) and the pulmonic valve (PV). LA = left atrium; LV = left ventricle; PA = pulmonary artery; RA = right atrium.
-
Ventricular Septal Defects. Echocardiogram from a child with a perimembranous ventricular septal defect (VSD). Note the defect at the 10 o'clock position in the parasternal short-axis view. AO = aortic root; LA = left atrium; LV = left ventricle; PA = pulmonary artery; RA = right atrium; RV = right ventricle.
-
Ventricular Septal Defects. Apical four-chamber views on computed tomography scanning. A: Image shows a large inlet defect. The defect is posterior and at the level of the atrioventricular valves. B: Image shows a small midmuscular ventricular septal defect. LA = left atrium; LV = left ventricle; PA = pulmonary artery; RA = right atrium; RV = right ventricle.
Tables
Syndrome |
CCVM (%) |
Type of CCVM |
Del 4q, 21, 32 |
60 |
Ventricular septal defect, atrial septal defect |
Del 5p |
30-60 |
Ventricular septal defect |
Trisomy 13 |
80 |
Atrial septal defect, ventricular septal defect, TOF |
Trisomy 18, Edwards syndrome |
100 |
Ventricular septal defect, TOF, double-outlet right ventricle (DORV) |
Trisomy 21, Down syndrome |
40-50 |
Ventricular septal defect, atrioventricular canal (AVC) |
Del 22q11, DiGeorge syndrome (single gene etiology, autosomal dominant) |
50 |
Truncus arteriosus, TOF, ventricular septal defect |





