Practice Essentials
Biliary atresia is characterized by obliteration or discontinuity of the extrahepatic biliary system, resulting in obstruction to bile flow. [1] Percutaneous liver biopsy is highly useful for evaluating neonatal cholestasis (see the image below). Surgical intervention is the only means available for a definitive diagnosis of biliary atresia (intraoperative cholangiography) and therapy (Kasai portoenterostomy).
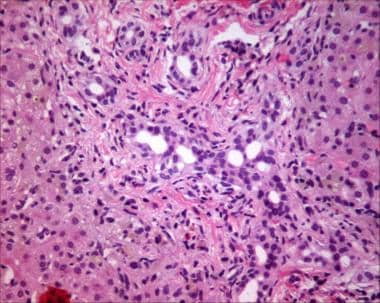 Bile ductular proliferation in liver biopsy specimen (hematoxylin and eosin stain) from patient with biliary atresia. Also note hepatocellular bile staining as a consequence of cholestasis.
Bile ductular proliferation in liver biopsy specimen (hematoxylin and eosin stain) from patient with biliary atresia. Also note hepatocellular bile staining as a consequence of cholestasis.
Signs and symptoms
Typical symptoms of neonatal cholestasis include the following:
-
Variable degrees of jaundice
-
Dark urine
-
Light stools
No physical findings are pathognomonic for biliary atresia; however, the following may be present:
-
Enlarged liver
-
Enlarged spleen
-
Direct hyperbilirubinemia (typically present from birth in the fetal/embryonic form of biliary atresia)
-
Physiologic jaundice that frequently merges into conjugated hyperbilirubinemia (in the postnatal form)
-
Midline liver (may be present in the fetal/neonatal form)
See Presentation for more detail.
Diagnosis
Laboratory studies
Laboratory studies that may be included in the workup include measurement of the following:
-
Serum bilirubin (total and direct)
-
Alkaline phosphatase
-
5' Nucleotidase
-
Gamma-glutamyl transpeptidase
-
Serum aminotransferases
-
Serum bile acids
-
Serum alpha1-antitrypsin with Pi typing
-
Sweat chloride
Imaging studies
In neonatal cholestasis syndromes, ultrasonography can exclude specific anomalies of the extrahepatic biliary system. Hepatobiliary scintiscanning is useful in evaluating infants with suspected biliary atresia.
Procedures
Percutaneous liver biopsy is widely regarded as the most valuable study for evaluating neonatal cholestasis. Intraoperative cholangiography definitively demonstrates the anatomy and patency of the extrahepatic biliary tract.
See Workup for more detail.
Treatment
No primary medical treatment is relevant in the management of extrahepatic biliary atresia. Once biliary atresia is suspected, surgical intervention is the only mechanism available for a definitive diagnosis (intraoperative cholangiography) and therapy (Kasai portoenterostomy).
See Treatment and Medication for more detail.
Background
Biliary atresia represents the most common surgically treatable cause of cholestasis encountered during the newborn period. If not surgically corrected, secondary biliary cirrhosis invariably results. Patients with biliary atresia can be subdivided into 2 distinct groups: isolated biliary atresia (postnatal form) and associated situs inversus or polysplenia/asplenia with or without other congenital anomalies (fetal/embryonic form).
Postnatal biliary atresia
Patients with isolated biliary atresia (postnatal form) account for 65-90% of cases. The neonatal type is characterized by a progressive inflammatory lesion, which suggests a role for infectious and/or toxic agents causing bile duct obliteration.
Embryonic biliary atresia
Patients with associated situs inversus or polysplenia/asplenia with or without other congenital anomalies (fetal/embryonic form) comprise 10-35% of cases. The fetal/embryonic form of atresia is associated with other congenital anomalies.
See the image below.
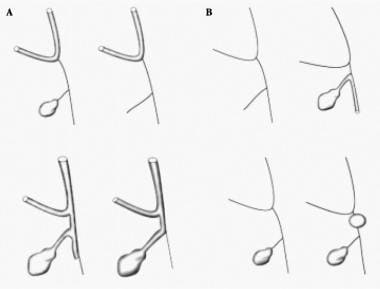
The pathology of the extrahepatic biliary system widely varies in these patients, and the following classification is based on the predominant site of atresia:
-
Type I involves obliteration of the common duct; the proximal ducts are patent
-
Type II is characterized by atresia of the hepatic duct, with cystic structures found in the porta hepatis
-
Type III (>90% of patients) involves atresia of the right and left hepatic ducts to the level of the porta hepatis. These variants should not be confused with intrahepatic biliary hypoplasia, which comprises a group of distinct and surgically noncorrectable disorders.
Pathophysiology
Although histopathologic features of biliary atresia have been extensively studied in surgical specimens from excised extrahepatic biliary systems of infants undergoing portoenterostomy, the pathogenesis of this disorder remains poorly understood. Early studies postulated a congenital malformation of the biliary ductular system. Problems of hepatobiliary ontogenesis are suggested by the fetal/embryonic form of atresia that is associated with other congenital anomalies. However, the more common neonatal type is characterized by a progressive inflammatory lesion, which suggests a role for infectious and/or toxic agents causing bile duct obliteration.
In type III, the most prevalent histopathological variant, the fibrous remnant demonstrates complete obliteration of at least a portion of the extrahepatic biliary system. Ducts within the liver, extending to the porta hepatis, are initially patent during the first few weeks of life but may progressively be destroyed. The same agent or agents that damaged the extrahepatic ducts may be causative, and the effects of retained toxins in bile are contributing factors.
Identification of active and progressive inflammation and destruction of the biliary system suggests that extrahepatic biliary atresia likely represents an acquired lesion. However, no single etiologic factor has been identified. Infectious agents seem to be the most plausible candidates, particularly in the isolated (neonatal) form of atresia. Several studies have identified elevated antibody titers to reovirus type 3 in patients with biliary atresia when compared with controls. Other viruses, including rotavirus and cytomegalovirus (CMV), have also been implicated.
Etiology
The disorder is rarely seen in infants who are stillborn or in premature infants, which supports a late gestational etiology. By contrast, infants with idiopathic neonatal hepatitis, which is the major differential diagnosis, are often preterm, small for gestational age, or both.
Infectious agents
No single agent has been identified as causative for biliary atresia, although the role of infecting organisms has been the most extensively studied.
Fischler et al reported cytomegalovirus (CMV) infection in almost 25% of affected infants in one study based on immunoglobulin M (IgM) serology. [2] Interestingly, an even higher frequency of CMV infection has been found by Chang et al in cases of idiopathic neonatal hepatitis, lending support to the concept that both disorders are ends of the same pathological spectrum, originally described by Landing as infantile obstructive cholangiopathy. [3]
Investigations of reovirus type 3 have yielded conflicting results. Wilson et al noted in one study that the virus damages the bile ducts and hepatocytes in mice, [4] whereas another study by Steele et al failed to demonstrate evidence of infection in infants with cholestasis. [5]
Other studies have examined the role of rotavirus groups A, B, and C and the common hepatitis viruses A, B, and C; however, no clear associations have been found. One study, using a murine model of biliary atresia induced by rhesus rotavirus, isolated trophism for the cholangiocyte to a specific genetic region. [6]
Genetic factors
The existence of the fetal/perinatal form of biliary atresia, frequently associated with other GI and cardiac anomalies, suggests the possibility of a disorder in ontogenesis. Studies have identified specific genetic mutations in mice with visceral heterotaxy and cardiac anomalies, defects similar to those found in conjunction with the fetal/perinatal form of biliary atresia.
Various genetic abnormalities, including deletion of the mouse c-jun gene (a proto-oncogene transcription factor) and mutations of homeobox transcription factor genes, are associated with hepatic and splenic defects. In a recent murine model, insufficient SOX17 expression in the gallbladder and bile duct epithelia resulted in biliary atresia. However, confirmation of a similar abnormality in human gene expression, and its potential etiopathogenetic role in this disorder, must await further studies. [7]
Other causes
Disorders of bile acid synthesis are part of the differential diagnosis of biliary atresia. In fact, bile acids almost certainly contribute to ongoing hepatocellular and bile ductular damage in infants with the disorder. Although associated defects in bile acid metabolism may hasten progression of liver disease, no primary role for bile acids in the development of biliary atresia has been identified.
Several investigators have studied the potential effects of other etiological agents, including teratogens and immunological factors. Again, no clear correlations with biliary atresia have been demonstrated.
Epidemiology
United States data
Individual studies suggest an overall incidence in the United States of 1 per 10,000-15,000 live births.
International data
The incidence of biliary atresia is highest in Asian populations, and it may be more common in Chinese infants compared with Japanese infants.
Race-, sex-, and age-related demographics
Incidence of biliary atresia is highest in Asian populations. The disorder also occurs in black infants, with an incidence approximately 2 times higher than that observed among white infants.
Extrahepatic biliary atresia is more common in females than in males.
Biliary atresia is a disorder unique to the neonatal period. Two presentations are described in this chapter (see Background). The fetal/perinatal form is evident within the first 2 weeks of life; the postnatal type presents in infants aged 2-8 weeks.
Prognosis
Data regarding outcome from centers worldwide widely vary. The initial success rate of Kasai portoenterostomy (for achieving bile flow) is 60-80%. Clearly, the most critical determinant of outcome remains age at the time of operation. Although individual centers have reported favorable surgical results in some infants older than 3 months, patients are significantly less likely to require early liver transplantation if the portoenterostomy is performed when they are younger than 10 weeks. In the postoperative period, the rate of decline in serum bilirubin levels directly correlates with a positive prognosis.
One study evaluated 244 infants who were enrolled in the prospective Childhood Liver Disease Research and Education Network and underwent Kasai portoenterostomy (KPE). The results noted that at 1 and 2 years post-KPE, the transplant-free survival rate was 53.7% and 46.7%, respectively. Risk of transplant/death was significantly lower in patients who achieved bile drainage within 3 months post-KPE, while it increased in patients with porta hepatis atresia, nonpatent common bile duct, biliary atresia splenic malformation syndrome, nodular liver appearance compared with firm, and age at KPE. No association with outcome was noted with gestational age, sex, race, ethnicity, or extent of porta hepatis dissection. [8]
Bile flow, even if achieved at surgery, may be inadequate in as many as one third of patients after the initial postoperative period. These children require early (< 2 y) liver transplantation. Practice guidelines for the evaluation of a patient for liver transplantation have been established by the American Association for the Study of Liver Diseases. [9] Factors that predict improved long-term outcome after Kasai portoenterostomy include the following:
-
Younger than 8 weeks at operation
-
Preoperative histology and ductal remnant size
-
Presence of bile in hepatic lobular zone 1
-
Absence of portal hypertension, cirrhosis, and associated anomalies
-
Experience of the surgical team
-
Postoperative clearing of jaundice
The following 3 categories of patients with extrahepatic biliary atresia should be considered for reexploration following a Kasai or modified Kasai portoenterostomy:
-
Infants who become jaundiced after an initial anicteric phase postoperatively
-
Infants with favorable hepatic and biliary duct remnant histology at initial operation, who do not successfully drain bile
-
Infants who may have had an inadequate initial surgery
Extrahepatic biliary atresia is the most common primary diagnosis in children requiring orthotopic liver transplantation (OLT), comprising more than 50% of patients with liver transplants in most series. [10] Overall, a review demonstrated that 66% of infants undergoing the Kasai procedure ultimately required OLT, including more than 50% of patients who initially achieved bile drainage.
In most series reported to date, the primary indications for OLT are the symptoms of end-stage liver disease and/or hepatic failure, including progressive cholestasis, recurrent cholangitis, poorly controlled portal hypertension, intractable ascites, decreased hepatic synthetic function (eg, hypoalbuminemia, coagulopathy unresponsive to vitamin K), and growth failure.
As long-term outcomes following OLT in children continue to improve (along with increased living donor availability) using split-liver grafts, application of this surgical modality for early treatment of biliary atresia will likely increase, certainly in patients with inadequate bile flow following portoenterostomy.
A study by LeeVan et al that included 626 children with biliary atresia reported that even though those that underwent primary liver transplant had a higher mortality rate after 3 months, those who underwent primary liver transplant had a reduced risk of long-term mortality than those managed with biliary-enteric drainage treatment after 6 months. [11]
Morbidity/mortality
Prior to the development of liver transplantation as a therapeutic option for children with end-stage liver disease, the long-term survival rate for infants with biliary atresia following portoenterostomy was 47-60% at 5 years and 25-35% at 10 years. In one third of all patients, bile flow is inadequate following surgery, and these children succumb to complications of biliary cirrhosis in the first few years of life unless liver transplantation is performed. Following portoenterostomy, complications include cholangitis (50%) and portal hypertension (>60%).
Hepatocellular carcinoma may be a risk for patients with cirrhosis and no clinical evidence of portal hypertension. Progressive fibrosis and biliary cirrhosis develop in children who do not drain bile. Thus, as discussed below (see Prognosis), liver transplantation may be the only option for long-term survival in most patients. [12]
A study by Leung et al evaluated 1,215 children (994 with biliary atresia, 221 with chronic liver disease) in the United Network for Organ Sharing registry data from 2003 to 2013 to investigate wait-list mortality associated risk factors, and outcomes of young children < 2 years of age. The study reported 12.4% wait-list mortality among this group and 8% posttransplant mortality. The wait-list mortality for children under 2 with chronic liver disease was 23.9% compared to 9.8% for bilateral atresia. The study also reported that the risk of death was 60% greater among patients with biliary atresia who did not have abdominal surgery (Kasai hepatoenterostomy) than among those with prior abdominal surgery. [13]
Complications
Complications following portoenterostomy in patients with biliary atresia include both acute and chronic problems. In the early postoperative phase, an unsuccessful anastomosis with failure to achieve adequate bile drainage is the most common complication. In this case, adequacy of bile flow may be predicted by the preoperative liver histology and the caliber of bile ductular remnants in the porta hepatis. In one third of all patients, bile flow is inadequate following surgery, and these children succumb to complications of biliary cirrhosis in the first few years of life unless orthotopic liver transplantation is performed.
Later in the course, complications related to progressive liver disease and portal hypertension occur in more than 60% of infants who achieved initial surgical success. Cholangitis develops in 50% of patients following portoenterostomy.
Hepatocellular carcinoma may be a risk for those patients with cirrhosis and no clinical evidence of portal hypertension. Progressive fibrosis and biliary cirrhosis develop in children who do not drain bile, and liver transplantation is the only option for long-term survival.
Detailed management of these complications is described in Histologic Findings, Medical Care, Consultations, Diet, and Medications.
-
Biliary atresia.
-
Bile ductular proliferation in liver biopsy specimen (hematoxylin and eosin stain) from patient with biliary atresia. Also note hepatocellular bile staining as a consequence of cholestasis.