Practice Essentials
Turner syndrome is one of the most common chromosomal abnormalities, occurring in approximately 1 in 2000 live-born female infants. [1, 2] Turner syndrome is caused by the absence of one set of genes from the short arm of one X chromosome.
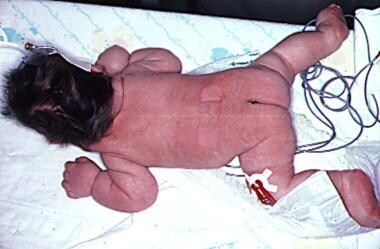 Generalized lymphedema is seen here in an infant with Turner syndrome. The loose skin folds around the neck will form a webbed neck later in life.
Generalized lymphedema is seen here in an infant with Turner syndrome. The loose skin folds around the neck will form a webbed neck later in life.
Signs and symptoms
At birth, girls with Turner syndrome may have swollen hands and feet because of lymphedema. In infants, the combination of dysplastic or hypoplastic nails and lymphedema gives a characteristic sausage-like appearance to the fingers and toes. Infants also have a higher incidence of congenital hip dislocation.
During childhood, girls with Turner syndrome usually present with short stature. [3] In older adolescents and adults, presenting symptoms usually involve issues of puberty and fertility as well as short stature.
Short stature
Growth rate in childhood is slightly slower; before age 11 years, some girls have height and growth rates that are well within the normal range, but heights are typically below the 50th percentile. [3]
The adolescent growth spurt is essentially absent.
Puberty
Adrenarche, the beginning of pubic hair growth, occurs at a normal age.
Breast development is absent when ovarian failure occurs before puberty.
Primary or secondary amenorrhea occurs with ovarian failure.
Other characteristic physical findings
These may include the following:
-
Dental: A high arched palate, sometimes with dental crowding or malocclusion
-
Nails: Hypoplastic or hyperconvex nails
-
Nevi: Excessive numbers of nevi, when compared to other family members
-
Webbed neck: A broad neck and a low or indistinct hairline
-
Cubitus valgus (increased carrying angle)
-
Madelung deformities of the wrist
-
Short fourth and fifth metacarpals and metatarsals
-
Shield chest: The chest appears to be broad with widely spaced nipples
-
Lymphedema
-
Eyes: Ptosis, strabismus, amblyopia, and cataracts; epicanthal folds can be present; red-green color blindness
-
Ears: Serous otitis media is more common [4] ; the auricles may be posteriorly rotated or low set; hearing loss due to otosclerosis is common in adults
-
GI bleeding: This is usually due to intestinal vascular malformations, but the incidence of Crohn disease and ulcerative colitis is also increased
-
Scoliosis: This occurs in 10% of adolescents with Turner syndrome and may contribute to short stature
-
Hypertension: May be caused by coarctation of the aorta or renal anomalies but often occur even in the absence of such findings
-
Cardiac murmurs: Cardiovascular malformations include hypoplastic left heart [5] , coarctation of the aorta, bicuspid aortic valve, and aortic dissection in adulthood
-
Thyroid: Hypothyroidism develops in 10-30% of patients [6] and is often associated with thyroid enlargement
-
Cutis laxa: Loose folds of skin, particularly in the neck, are signs in newborns; this is a result of resolving lymphedema and occasionally is observed after infancy
Diagnosis
Prenatal
On fetal ultrasonography, Turner syndrome is suggested by the presence of a nuchal cystic hygroma, [7, 8] horseshoe kidney, left-sided cardiac anomalies, or nonimmune fetal hydrops.
Turner syndrome may be prenatally diagnosed by amniocentesis or chorionic villous sampling.
Noninvasive prenatal testing of maternal blood can be used to screen for Turner syndrome with great sensitivity and specificity. [9]
Karyotyping
A standard 30-cell karyotype analysis is required for diagnosis of Turner syndrome, to exclude mosaicism. [10]
Diagnosis is confirmed by the presence of a 45,X cell line or a cell line with deletion of the short arm of the X chromosome (Xp deletion).
The buccal smear for Barr bodies is obsolete.
A male phenotype excludes the diagnosis, regardless of karyotype. [11]
Patients with Turner syndrome should be investigated for the presence of Y chromosomal material using a Y-centromeric probe.
Laboratory studies
Gonadotropins: Both LH and FSH may be elevated in untreated patients younger than 4 years; they are later suppressed to normal or near-normal levels, only to rise to menopausal levels after age 10 years. [12]
Thyroid function tests: Because of the high prevalence of hypothyroidism in Turner syndrome, [6] obtain thyroid function tests at diagnosis; repeat TSH measurements every year in early childrhood or if symptoms develop, because hypothyroidism may develop at a later age.
Celiac screening: There is an increased risk for autoimmune diseases. Screening should be done at age 2 and every 2 years afterward in childhood. [13]
Glucose metabolism: Abnormalities of glucose metabolism, including overt diabetes mellitus, are more common than in unaffected children; screening for diabetes mellitus is best performed by obtaining a hemoglobin A1c.
Renal studies
At diagnosis, perform ultrasonography of the kidneys and renal collecting system.
Annual urine cultures and measurement of BUN and creatinine levels are recommended for those patients with abnormalities of the renal collecting system that predispose to obstruction.
Cardiovascular studies
Perform echocardiography and/or MRI of the heart and aorta upon diagnosis.
Evaluate 4-limb blood pressures, because of the high incidence of coarctation of the aorta.
Audiology
Infants diagnosed at birth should have a hearing assessment in the nursery.
Formal hearing assessment is recommended at age 1 year and before entering school.
Formal re-evaluation every 3 years has been recommended [13] ; more frequent testing is needed in children with repeated otitis media.
Adults should have a hearing evaluation every 5 years [13]
Management
Patients with Turner syndrome require screening for commonly associated chronic diseases. Early preventive care and treatment are also essential. [14]
Growth hormone therapy
In childhood, growth hormone therapy is standard to prevent short stature as an adult. [15, 16, 17]
Ideally treatment should be initiated in early childhood; taller adult heights occur with the longest treatment durations before the start of puberty.
Growth hormone may have long-term favorable effects on lipids, even after it is discontinued. [18]
Sex hormone replacement therapy
Estrogen replacement therapy is usually required, but starting too early or using doses that are too high can compromise adult height.
Estrogen is usually started at age 11-12 years if gonadotropins are elevated. [13] Delaying onset of puberty can compromise social and physical development.
Continuous low-dose estrogens can be cycled in a 3-weeks on, 1-week off regimen; progestin can be added later approximately 2 years after starting estrogen therapy.
Some authors believe that conjugated estrogens are contraindicated in pediatric patients. [10]
Transdermal estrogens are associated with physiologic estrogen levels [19] and may be the preferred treatment, if tolerated. [10]
Androgen replacement therapy is not the standard of care, [10] but may have favorable effects. [20]
Consultations
These include the following:
-
Endocrinologist
-
Cardiologist
-
Nephrologist or urologist
-
Psychologist
-
Genetics
Diet
Both short stature and ovarian failure are risk factors for osteoporosis, and care should be taken to ensure adequate daily intake of calcium (1.0-1.5 g) and vitamin D (at least 400 IU).
Patients should avoid obesity, which increases already high risks of hypertension and insulin resistance.
Background
In 1938, Henry Turner first described Turner syndrome, which is one of the most common chromosomal abnormalities. [1] More than 95% of adult women with Turner syndrome exhibit short stature and infertility. Examples of manifestations of Turner syndrome are shown in the images below.
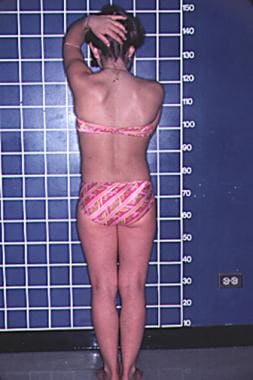 A patient with Turner syndrome is shown. This posterior view shows a low hairline and a shield-shaped chest. Note the narrow hip development.
A patient with Turner syndrome is shown. This posterior view shows a low hairline and a shield-shaped chest. Note the narrow hip development.
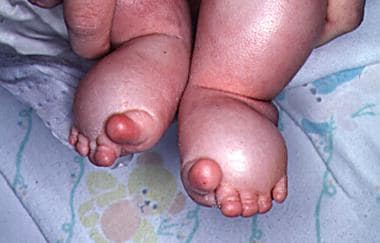 Lymphedema of the feet in an infant is shown. The toes have the characteristic sausagelike appearance.
Lymphedema of the feet in an infant is shown. The toes have the characteristic sausagelike appearance.

Pathophysiology
Turner syndrome is caused by the absence of one set of genes from the short arm of one X chromosome. In patients with 45,X karyotype, about two thirds are missing the paternal X chromosome. In addition to monosomy X, a similar clinical picture is found with a 46,XXiq karyotype and in some individuals with mosaic karyotypes. A deletion of the SHOX gene can cause a similar skeletal phenotype known as Leri-Weill dyschondrosteosis.
Frequency
United States
The frequency of Turner syndrome is approximately 1 in 2000 live-born female infants. [2] As many as 15% of spontaneous abortions have a 45,X karyotype. Interestingly, 99% of conceptions with 45,X karyotypes spontaneously abort. [21]
International
The incidence is the same as in the United States. No known ethnic or racial factors influence frequency.
Mortality/Morbidity
Mortality associated with Turner syndrome may be increased in the neonatal period because of hypoplastic left heart [5] and coarctation of the aorta and in adulthood because of cardiovascular disease, particularly aortic dissection.Obesity, with associated diabetes mellitus and hypertension, can also contribute to early mortality. Limited epidemiologic studies suggest that life expectancy is reduced by about 10 years. Osteoporosis is common.
Although congenital cardiovascular malformations and aortic dilatation are common among patients with Turner syndrome, they are often undiagnosed until later in life, pointing to the need for a more systematic approach to cardiovascular monitoring. [22]
Renal anomalies found in some individuals may cause a predisposition to urinary tract infections or hypertension. Even in the absence of cardiac or renal anomalies, patients are prone to develop hypertension.
Individuals with mitral valve disease or aortic valve disease require subacute bacterial endocarditis (SBE) prophylaxis.
In a Danish study, the overall risk of autoimmune disease among women with Turner syndrome was twice that among Danish women in general. [23] For individual diseases, associations were strongest for Autoimmune thyroiditis, a condition more common in females, and type 1 diabetes mellitus.
Race
No racial or ethnic predilections are known.
Sex
Turner syndrome only occurs in females. [11] Noonan syndrome, sometimes inappropriately called male Turner syndrome, can occur in males or females. It is an autosomal dominant genetic disorder and is not a chromosomal disorder. It is unrelated to Turner syndrome.
Age
As a chromosomal disorder, Turner syndrome is present at conception or following the first cell division and remains throughout life. Gonadotropin levels, particularly follicle-stimulating hormone (FSH) levels, may be elevated at birth, although not reliably enough for use in neonatal screening. [24] They are gradually suppressed by about age 4 years, only to rise to menopausal levels after age 10 years. [12]
Prognosis
Overall prognosis for patients with Turner syndrome is good.
Even with growth hormone therapy, most individuals are shorter than average. [25]
Turner syndrome is not a cause of intellectual disability. [26] They are more likely to be employed than other adult women. [27]
Life expectancy is slightly shorter than average but may be improved by attention to associated chronic illnesses, such as obesity and hypertension. [28]
Almost all individuals are infertile, but pregnancy with donor embryos is possible. [29]
Patient Education
For patient education resources, see the Thyroid and Metabolism Center, the Women's Health Center, and the Amenorrhea article.
-
A patient with Turner syndrome is shown. This posterior view shows a low hairline and a shield-shaped chest. Note the narrow hip development.
-
Lymphedema of the feet in an infant is shown. The toes have the characteristic sausagelike appearance.
-
Hyperconvex nails in Turner syndrome. Note U-shaped cross section.
-
Generalized lymphedema is seen here in an infant with Turner syndrome. The loose skin folds around the neck will form a webbed neck later in life.








