Practice Essentials
The thalassemias are a group of inherited disorders in which globin chain production is reduced or absent. Beta thalassemia results from beta-globin gene mutations that impair beta-globin chain synthesis. [1] Clinical severity forms the basis of beta thalassemia classification, as follows [1] :
-
Beta thalassemia major - The severest type of beta thalassemia, with patients suffering from severe anemia and transfusion dependency
-
Beta thalassemia intermedia - Sporadic or no transfusions are required for anemia
-
Beta thalassemia minima - Also called beta thalassemia trait, this form is usually asymptomatic
This article will focus on beta thalassemia intermedia.
In patients with beta thalassemia intermedia, anemia is present but individuals are not transfusion dependent. A clinically heterogeneous group, patients with this disease can have symptoms that range from mild anemia, with only a rare need for transfusions, to chronic hemolysis and the development later in life of transfusion dependence.
The initial workup for a patient with suspected thalassemia should include a complete blood count (CBC), review of the blood smear, and iron studies. Thalassemia intermedia therapy is aimed not only at treating the anemia, but also at addressing iron overload and inhibiting the clinical consequences of ineffective erythropoiesis.
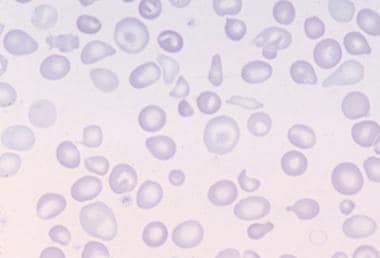
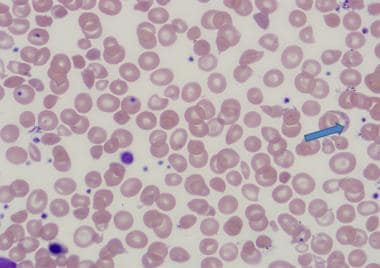
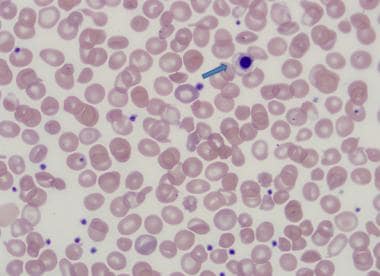
The following are histologic images from patients with thalassemia intermedia.
See also Beta Thalassemia, Alpha Thalassemia, Thalassemia, Alpha, Pediatric Thalassemia, Thalassemia Imaging, and Anemia.
The initial workup for a patient with suspected thalassemia should include a complete blood count (CBC), review of the blood smear, and iron studies. Thalassemia intermedia therapy is aimed not only at treating the anemia, but also at addressing iron overload and inhibiting the clinical consequences of ineffective erythropoiesis.
Signs and symptoms of thalassemia
Despite maintaining a level of hemoglobin sufficient for tissue oxygenation, patients with thalassemia can develop a range of complications due to longstanding hemolytic anemia and ineffective erythropoiesis, including jaundice, splenomegaly, bone deformities, osteoporosis, fractures, growth retardation, extramedullary hematopoietic pseudotumors, [2] pulmonary hypertension, thromboembolism, [3] iron overload, and skin ulcers.
Workup in thalassemia
Laboratory studies in the workup of thalassemia include the following:
-
CBC - Thalassemia involves microcytic anemia with a high red blood cell count, with the high count possibly helping to distinguish between thalassemia and iron deficiency [1]
-
Peripheral smear - Review of the peripheral smear reveals hypochromasia and microcytosis; more severely affected patients can demonstrate poikilocytosis, with target cells, teardrop cells, and cell fragments [1]
-
Iron studies - Iron studies should be obtained to differentiate iron deficiency anemia from thalassemia and also to monitor for ongoing iron overload in patients with an established diagnosis of thalassemia; in thalassemia intermedia, iron studies reveal a normal to high ferritin level, elevated serum iron, and elevated transferrin saturation [1]
Management
Patients undergo transfusions as needed, most often in response to stressors such as illness, pregnancy, surgery, and periods of rapid growth. [1] In later life, some patients become transfusion dependent.
Even without chronic transfusion, iron overload can develop in the presence of beta thalassemia intermedia consequent to increased iron absorption from the gut. [1] Available chelation therapy agents include deferoxamine (parenterally administered) and deferiprone and deferasirox (both orally administered). With the introduction of deferiprone and deferasirox, deferoxamine, due to its cumbersome administration schedule, fell out of favor in North America and Europe.
Prompt evaluation for paraspinal extramedullary hematopoietic pseudotumors should conducted, via spinal magnetic resonance imaging (MRI), in patients with beta thalassemia in whom the symptoms and signs of spinal cord compression are present. [4] Stimulation of fetal hemoglobin by way of a short course of hypertransfusion and hydroxyurea can be used to treat mild symptoms. [1] Low-dose radiotherapy and steroids can also be considered. Laminectomy may be required to address severe symptoms that fail to respond to medical therapy. [1]
Splenectomy should be reserved for patients with massive splenomegaly, worsening pancytopenia, or hypersplenism. Among its many risks, the greatest dangers related to splenectomy are postsplenectomy sepsis and thrombosis, with these being particular hazards in patients with beta thalassemia intermedia.
In patients with severe thalassemia intermedia who require aggressive therapy to sustain life, bone marrow transplantation, similar to that performed in patients with thalassemia major, is a reasonable alternative to transfusion and chelation if a matched sibling donor is available.
Pathophysiology
Normal hemoglobin is a tetramer composed of two alpha-like and two beta-like globin chains. The predominant hemoglobin at birth is hemoglobin F (HbF) with the structure α2γ2. A switch from γ to beta globulin synthesis occurs such that by 4-6 months of life, the main circulating hemoglobin is hemoglobin A (HbA), with a structure of α2β2.
The beta-globin gene cluster, found on the short arm of chromosome 11, can be affected by over 200 known (primarily point) mutations that give rise to beta thalassemia. The following terminology is commonly used:
-
β 0 - Mutations causing a complete absence of beta-globin chains
-
β + - Mutations resulting in a variable decrease in the production of beta globin
Patients with beta thalassemia intermedia are considered typically to have two β+ mutations. However, owing to the heterogeneity of the beta-globin mutations and the multitude of disease modifiers, difficulties remain regarding the prediction of phenotype from genotype. Thus, transfusion independence is still the best means of defining beta thalassemia intermedia. [1]
In beta thalassemia, beta-globin gene mutation leads to the decrease or absence of beta-globin chain production. [1] The two main implications of this defect are as follows:
-
Decreased hemoglobin production
-
Imbalanced alpha-chain–to–beta-chain ratio
Decreased hemoglobin production
Impairment in the production of functional hemoglobin tetramers takes the form of microcytosis and hypochromasia, as readily demonstrated in peripheral smears from patients with beta thalassemia intermedia.
Imbalanced ratio of alpha-to-beta chains
Another consequence of impaired beta-globin synthesis is the imbalanced ratio of alpha to beta chains. It is essential to understand that the clinical severity of beta thalassemia arises not from the absolute deficit in beta-globin production but from the degree of globin chain imbalance. [1] The excess alpha chains precipitate, the result being that red blood cell precursors in the bone marrow and circulation are destroyed, leading, via ineffective erythropoiesis and hemolysis, to anemia. This erythropoiesis/hemolysis inefficacy also results in iron overload, erythroid marrow expansion, and a hypercoagulable state. Iron overload can develop even in non–transfusion-dependent patients owing to inappropriately low hepcidin levels, which result in increased intestinal iron absorption.
The heterogeneity of the beta thalassemia intermedia phenotype can also be attributed to other variables that may affect the alpha-globin–to–beta-globin ratio and, thus, disease severity, including the following:
-
Coinherited genetic determinants, such as differential molecular forms of alpha thalassemia, can reduce the alpha-chain/beta-chain production imbalance [1]
-
HbF levels will rise in association with gamma-chain production increases; such is the case with beta-/delta-deletion mutations, which, in association with a beta thalassemia gene mutation, lead to thalassemia intermedia via a combined heterozygous condition [1]
-
Patients with HbE/beta thalassemia (in which HbE interacts with beta thalassemia) exhibit the clinical course of thalassemia intermedia
Prognosis
Being clinically heterogenous, beta thalassemia intermedia can range from a manifestation in which patients rarely, if ever, require transfusion to one in which individuals have chronic hemolytic anemia and in later life become transfusion dependent. [1]
Although these patients sustain hemoglobin levels adequate for survival, the underlying ineffective erythropoiesis can fuel various complications, including iron overload, extramedullary hematopoiesis, hemolysis, and hypercoagulability. (Presentation/Clinical Manifestations)
-
Peripheral blood film in thalassemia intermedia.
-
Basophilic stippling in thalassemia intermedia.
-
Nucleated red blood cell in thalassemia intermedia.








