Practice Essentials
Of genetic disorders worldwide, thalassemia syndromes are among the most common. Normal adult hemoglobin produced after birth (hemoglobin A [HbA]) consists of a heme molecule linked to two α-globin and two β-globin chains (α2β2), with α-globin chain production dependent on four genes on chromosome 16, and β-globin chain production arising from two genes on chromosome 11. Deletions or mutations of one or more of these genes so that the rate of production of α- or β-globin chains is reduced results in alpha thalassemia or beta thalassemia, respectively. Thalassemia is usually asymptomatic in carriers, or presents with anemia of varying degrees in patients in whom globin-chain production is more severely impaired. [1]
Patients with alpha-thalassemia trait or beta-thalassemia trait are asymptomatic but have mild microcytic hypochromic anemia, which often goes undiagnosed or is confused with iron deficiency anemia. Recognizing the possibility of thalassemia trait by taking a complete family history and appropriate testing is important in making an accurate diagnosis. Individuals with thalassemia trait may be at risk of having a severely affected child and should be referred for genetic counseling when appropriate. [2] Similarly, the birth of a child with severe thalassemia is a trigger for genetic counseling and future prenatal testing.
Patients with severe beta thalassemia are dependent on red cell transfusions either regularly (thalassemia major) or intermittently (thalassemia intermedia). Regardless of their transfusion needs, such patients should be followed at a thalassemia comprehensive care center under the care of a hematologist, so that they can be monitored for short- and long-term complications of chronic transfusions, including iron overload with cardiac and liver damage, as well as for growth and endocrine issues, bone pathology, and infertility. Curative therapy such as bone marrow transplantation may be an option for some patients, and novel agents, as well as gene therapy, are in the pipeline. [3, 4, 5]
Patients with severe alpha thalassemia requiring red cell transfusion (HbH disease) should be monitored closely in a similar fashion. Recognizing that nonimmune hydrops fetalis in mothers of Southeast Asian origin can be due to severe alpha thalassemia is important for genetic counseling and future prenatal testing. Rarely, patients with Hb Bart hydrops fetalis have been salvaged with intrauterine transfusions, but there is considerable morbidity, and this is not the standard of care. [6]
Presentation in pediatric thalassemia
Severe forms of beta thalassemia are characterized by the following physical findings, particularly if the patient is inadequately transfused:
-
Severe pallor, scleral icterus
-
Enlarged abdomen due to hepatosplenomegaly
-
Severe bony changes due to ineffective erythroid production (eg, frontal bossing, prominent facial bones, dental malocclusion)
-
Neuropathy/paralysis due to extramedullary hematopiesis
-
Growth retardation and short stature
Patients with alpha thalassemia, even those with a severe form (having lost 3 out of 4 genes), will have findings of mild to moderate hemolytic anemia, as follows:
-
Pallor, scleral icterus
-
Splenomegaly (hepatomegaly is less common)
-
Absence of bony deformities
Workup in pediatric thalassemia
Complete blood count (CBC) results and red cell indices, along with peripheral blood film examination outcomes, are usually sufficient to suspect a diagnosis of thalassemia. Hb electrophoresis can usually confirm the diagnosis of beta thalassemia, HbH disease, and HbE/β-thalassemia.
Globin chain synthesis, which was once used in postnatal diagnosis, has also been used on fetal cells obtained by fetoscopy to screen the fetus for thalassemia.
Since polymerase chain reaction (PCR) assay techniques became available, several new methods have come into use to identify affected babies or carrier individuals accurately and quickly. Moreover, the sensitivity of next-generation sequencing (NGS) has allowed noninvasive screening to be done on fetal DNA obtained from maternal plasma.
Management
Splenectomy is the principal surgical procedure used for some patients with thalassemia. However, with reports made of venous thromboembolic events (VTEs) after splenectomy, one should carefully consider the benefits and risks before splenectomy is advocated.
Patients typically receive PRBC transfusions (up to 20 mL/kg) every 3-4 weeks, with clinicians aiming for a 9-10 g/dL hemoglobin level prior to the next transfusion. In some patients, shorter intervals between transfusions may be beneficial. [7]
Routine administration of iron chelation is essential to avoid transfusion-related iron overload and multiorgan (especially cardiac and liver) toxicity.
In 2019, the European Union conditionally approved the use of betibeglogene autotemcel (Zynteglo), the first gene therapy for the treatment of transfusion-dependent beta thalassemia. [5, 8, 9] The US Food and Drug Administration (FDA) approved the betibeglogene autotemcel in August 2022. A second gene therapy, exagamglogene autotemcel (Casgevy), was approved by the FDA for transfusion-dependent beta thalassemia in 2024.
Background
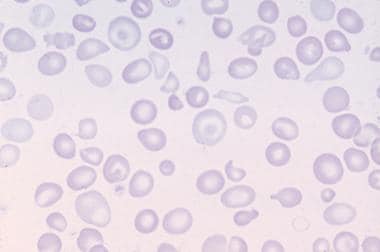
Beta thalassemia was the first described in 1925, by Thomas Cooley, a Detroit pediatrician, who reported on children of Italian origin who presented with severe microcytic anemia and other red cell abnormalities (see image below), enlarged liver and spleen, and skull and bony abnormalities. Because of the patients’ ethnic origin, “Cooley's anemia” was later renamed thalassemia (thalassa in Greek means "great sea" or Mediterranean). [10] In 1959, Ingram and Stratton postulated that decrease in β-globin or α-globin production led to a transfusion-dependent anemia, and in the latter case it resulted in HbH (β4) disease, which we now recognize as severe alpha thalassemia. [11] Three years later, Lie-injo Luan Eng, an Indonesian pathologist, described a stillbirth with Hb Bart hydrops fetalis, the most severe manifestation of alpha thalassemia. [12] We now recognize a number of thalassemia syndromes and have a better understanding of the underlying pathophysiology.
Pathophysiology
HbA, or α2β2, consists of heme combined with two α-globin and two β-globin chains. On chromosome 16, each DNA strand has two α-globin genes, whereas chromosome 11 has a single pair of β-globin genes. Nevertheless, the globin-chain output of these genes is closely matched to effectively produce HbA. In the thalassemia syndromes, mutations affecting either gene affect this balanced production of α-globin and β-globin chains, resulting in decreased hemoglobin and varying degrees of anemia. [8]
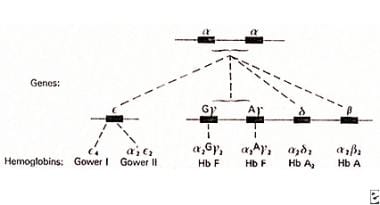 Alpha chain genes in duplication on chromosome 16 pairing with non-alpha chains to produce various normal hemoglobins.
Alpha chain genes in duplication on chromosome 16 pairing with non-alpha chains to produce various normal hemoglobins.
During fetal development, globin-producing genes are switched on and off to produce different hemoglobins (see figure above). The γ-globin gene is switched on for the majority of the time in utero, producing fetal hemoglobin (HbF), or α2γ2. After birth, this changes in a few months to adult hemoglobin (HbA), or α2β2, with small amounts of HbA2, or α2δ2 (δ-globin production being physiologically impaired). Not shown in the figure are genes active only in early embryonic life: ζ-globin, which precedes α-globin, combines with γ-globin to produce Hb Portland (ζ2γ2), and ε-globin, which precedes γ-globin and forms Hb Gower (ζ2ε2, α2ε2), a hemoglobin of no clinical significance. Upstream of these globin gene clusters are regulatory elements that help to switch globin gene activity on and off.
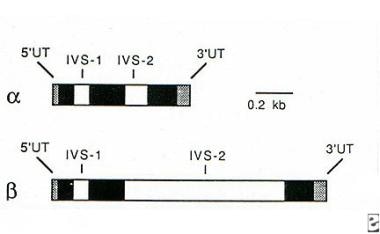
Each globin gene consists of three coding exons and two noncoding introns, or intervening sequences (IVS) (see image below). This knowledge is especially relevant for beta thalassemia, in which over 200 point mutations can impair β-globin synthesis, and the location of these mutations is often described in terms of relationship to IVS-1 or IVS-2.
The synthesis of globin proteins at a molecular level is well understood, but to summarize: when a globin gene is transcribed, a messenger RNA (mRNA) precursor corresponding to one of the gene's DNA strands is synthesized. This contains exons and introns, so the mRNA is then processed by eliminating the introns and splicing together the exons, which requires recognition of specific GT/AG base pairs at the “splice sites.” The 5’ and 3’ ends of the mRNA are then modified, and the processed mRNA moves from the nucleus to the cytoplasm. In conjunction with a ribosome, this mRNA now acts as a template for a series of transfer RNA (tRNA) molecules, each bringing an amino acid based on codon-anticodon base pairing. This translation process assembles a string of amino acids into a peptide, which continues until a specific “stop” codon is reached. The completed globin chain then drops off the ribosome-mRNA complex and joins a heme molecule and 3 other globin chains to form a hemoglobin molecule.
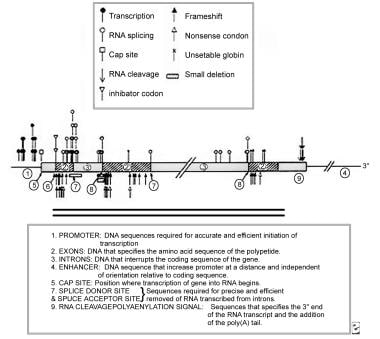
Beta thalassemia is usually caused by mutations affecting a single nucleotide substitution, which can impact each step of this process (see figure below). Authors refer to severe mutations, with complete absence of β-globin production, as β0 mutations, and refer to less severe mutations as β+ mutations. It is important to keep in mind that the severity of the mutation may not always correlate with the clinical picture. [13] Splice-site mutations, which are especially common, change the critical GT/AG bases around the splice site (eg, IVS1-1 G>T), rendering the splice site unrecognizable by the normal splicing process. In a “nonsense” mutation, a single base change in the exon generates a stop codon in the mRNA, resulting in premature termination of the globin chain. In a “frameshift” mutation, one or more bases on the exon are lost or inserted, resulting in a change in the reading frame of the genetic code or the production of a new stop codon. Mutations in exons may also activate a cryptic splice site, as in HbE, in which a mutation at codon 26 (G>A) results in alternate splicing, reducing the amount of β-globin production (similar to β+ thalassemia). [14] Rarely, deletions, rather than point mutations, have been described; in Hb Lepore, a deletion leads to a fused δ/β gene, under the control of the δ-globin gene promoter, which is weak (so that mild beta-thalassemia–like behavior results).
Alpha thalassemia results from the deletion of one or both of the α-globin genes on the same DNA strand, with more than 35 such deletions described. Severe α0-thalassemia carriers are represented as αα/- - to show the absence of both α-globin genes on the same strand, which is due to large deletions such as --SEA, --MED, --FIL, --THAI, or --20.2. This is a common finding in alpha-thalassemia carriers from Southeast Asia, southern China, or the Middle East, who are therefore at risk of having a stillbirth with Hb Bart hydrops fetalis (- -/- -). In contrast, in α+-thalassemia, smaller deletions of 3.7 or 4.2 kb from the α-globin gene (α-3.7 or α-4.2) remove only a single gene, so that alpha-thalassemia carriers from Africa (- α/- α) are not at risk within their own community. If an α+-thalassemia carrier and an α0-thalassemia carrier have a child, there is a risk of HbH disease (- -/- α). In addition, point mutations can occur so that in Hb Constant Spring (αCSα) or Hb Quong Sze the stop codon for the α-globin gene is affected, generating a long, unstable globin chain. Such nondeletional alpha-thalassemia mutations interfere with the remaining α-globin gene production on the same DNA strand, so that - -/ αCSα causes HbH disease that is more severe than that resulting from gene deletion. [15]
Mortality/Morbidity
In patients with thalassemia, mortality and morbidity vary according to the severity of the disease and the quality of care provided. Severe cases of beta-thalassemia major are transfusion-dependent, and chronic iron overload or undertransfusion can lead to cardiac failure, liver disease, chronic or acute infection, and other complications. Even patients receiving well-designed treatment regimens may be at risk for a variety of complications. [16]
Hb Bart hydrops fetalis is lethal, and fetuses are stillborn with severe anemia, which is traumatic to the mother and family. Intrauterine blood transfusions have salvaged some patients in specialized centers, but there is considerable morbidity, and this is not a standard of care or an option for the vast majority of patients affected worldwide. [15]
Patients with HbH disease usually have mild hemolytic anemia requiring only occasional blood transfusions, but some patients who have co-inherited nondeletional mutations such as Hb Constant Spring or Hb Quong Sze have more severe, transfusion-dependent anemia. These patients may require splenectomy, and morbidity is very similar to patients with beta-thalassemia intermedia. [17]
Epidemiology
Thalassemias are encountered among all ethnic groups and in almost every country around the world, with 15 million people worldwide having clinical thalassemic disorders. There is a wide variation in the prevalence rate for alpha and beta thalassemia in different parts of the world. In areas endemic for beta thalassemia, such as the Mediterranean countries and islands, the Middle East, and the Indian subcontinent, the carrier rate is 10-15%. The lack of systematic preventive measures in lower-income countries, means that in India alone, 10,000 new beta-thalassemia patients are added to the population each year. [18] Regions impacted by severe alpha thalassemia are Southeast Asia, the Middle East, and southern China, where the carrier rate exceeds 5%, with the rate approaching 5% in Thailand. In southern China, there are 2-3 times more fetuses afflicted with the lethal Hb Bart hydrops fetalis than with severe beta thalassemia. [19, 15]
In the United States, a diverse immigrant population has meant that thalassemia can occur in any part of the country. However, the number of patients with severe alpha or beta thalassemia is limited, so finding more than 2-5 patients in any pediatric hematology center is unusual (except in a few referral centers). Nonetheless, alpha thalassemia is increasingly prevalent in the United States and accounts for more than 50% of non–transfusion-dependent thalassemia (unpublished data, courtesy of Janet Kwiatkowski). The prevalence of alpha and beta thalassemia, as well as of HbE/β-thalassemia, is increasing in California, due to a high prevalence of individuals of Asian origin, and the cord-blood screening program for detection of hemoglobinopathy there annually detects 10-14 cases of beta-thalassemia major and HbE/β-thalassemia and 40 cases of HbH disease. [20]
Prognosis
Patients with alpha- or beta-thalassemia trait have a normal lifespan, while Hb Bart hydrops fetalis (homozygous α0 thalassemia) is lethal in utero. With regular transfusions of red cells and comprehensive care, including aggressive iron chelation, life expectancy in birth cohorts with severe beta thalassemia has been found to extend into the fourth decade and beyond. Patients with HbH disease or beta-thalassemia intermedia can be expected to survive at least as long, depending on transfusion needs and availability of care. [7]
-
Alpha chain genes in duplication on chromosome 16 pairing with non-alpha chains to produce various normal hemoglobins.
-
Alpha and beta globin genes (chromosomes 16 and 11, respectively).
-
Various mutations in the beta gene that result in beta thalassemia.
-
Supra vital stain in hemoglobin H disease that reveals Heinz bodies (golf ball appearance).
-
Peripheral blood film in Cooley anemia.
-
Peripheral blood film in thalassemia minor.
-
Peripheral blood film in hemoglobin H disease in a newborn.
-
Peripheral blood in iron deficiency anemia.
-
The classic "hair on end" appearance on plain skull radiographs of a patient with Cooley anemia.
-
Excessive iron in a bone marrow preparation.








