Background
Behçet syndrome is a multisystem disease of unknown etiology probably first described by Hippocrates in the 5th century. The syndrome carries the name of the Turkish dermatologist Hulusi Behçet, who, in 1937, described a syndrome of recurrent aphthous ulcers, genital ulcerations, and uveitis leading to blindness. Although the cause of the disease is still unknown, it has become recognized as a multisystemic inflammatory disease with a heterogeneity of clinical manifestations. Complicating matters, clinical presentations vary among geographical regions and manifestations tend to cluster together. See the images below.
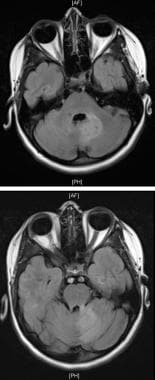 MRI, T2-weighted images of brainstem involvement with meningoencephalitis in an 11-year-old girl with neurologic Behçet syndrome.
MRI, T2-weighted images of brainstem involvement with meningoencephalitis in an 11-year-old girl with neurologic Behçet syndrome.
Pathophysiology
Behçet syndrome is characterized by recurrent aphthous ulcers, genital ulcers, and uveitis or retinal vasculitis. Other manifestations of the disease include skin lesions, arthritis, GI lesions, CNS involvement, and vascular lesions, including aneurysms and thrombosis. In Behçet syndrome, the basic lesion is vasculitis. Biopsies have shown vasculitis near affected lesions, including the oral and genital ulcers and lesions of the CNS and the eyes; large vessels are affected by a vasculitis of the vasa vasorum. Vascular injuries may be superimposed on the hypercoagulability observed in some patients.
Neutrophilic hyperfunction is observed in patients with Behçet syndrome with neutrophilic infiltration of skin at the site of a prick with a sterile needle (the pathergy test). Lymphocyte function has also been reported as abnormal, with a clonal expansion of autoreactive T cells.
Results of recent genome-wide association studies (GWASs) confirm the association with HLA-B51, although there is currently not a diagnostic or prognostic role for the presence of HLA-B51. These GWASs demonstrate that the most common non–HLA association is with the interleukin (IL)–10 and IL23R loci and underline the essential role of the IL-10 and IL23/17 pathways in the pathogenesis of Behçet syndrome. [1] IL-6 has been shown to be elevated and IL-10 decreased in patients with Behçet syndrome compared with controls, and IL-6 is elevated in those with active disease. [2]
Epidemiology
Frequency
United States
Frequency data for Behçet syndrome should be considered suspect because of problems with case ascertainment. [3] This problem is inherent in any disease where no specific diagnostic test exists and only a set of clinical criteria is used for diagnosis. Figures available from Olmstead County, Minnesota reveal prevalence in this community to be 5 cases per 100,000 persons. Other estimates of prevalence vary from 0.12-0.33 cases per 100,000 persons.
International
Behçet syndrome is thought to be more common along the ancient Silk Road, extending from Asia to the Mediterranean. Estimates from Turkey vary from 80-370 cases per 100,000 population, whereas prevalence estimates from Japan, Korea, China, and the Middle East vary from 13-20 cases per 100,000 population. In northern Spain, prevalence has been reported as 0.66 cases per 100,000 population, whereas estimates from Germany are 2.26 cases per 100,000 population.
A review by Leccese et al reported similarities in Behcet’s prevalence between regions with immigrant populations and originating countries, however, symptoms are less severe. [4]
Mortality/Morbidity
See the list below:
-
There is a wide range of phenotypic variations in Behçet syndrome, which may be due to genetic and ethnic differences.
-
Ocular: Uveitis occurs in 60-80% of patients. Retinal arterial and venous lesions are prognostic indicators for blindness, which is a major complication of Behçet syndrome. In Middle Eastern populations, the mean time from onset of disease to blindness is 3-4 years in untreated patients or in those treated only with corticosteroids.
-
CNS: Neurologic involvement is one of the most serious manifestations of Behçet syndrome, occurring in 10-30% of patients and carrying a poor prognosis. Manifestations include meningitis or meningoencephalitis; psychiatric symptoms, including personality changes; neurological deficits, including hemiparesis; and brainstem symptoms. Neurological deficits may be progressive, with 30% of those patients with neurologic manifestations eventually developing dementia. Compared with adults, children tend to have dural sinus thrombosis more often than parenchymal disease. [5]
-
Vascular: Vascular involvement in Behçet syndrome is unique in that it involves both the arterial and venous systems. Vascular complications, which occur in 7-40% of patients, include venous and arterial thromboses, vessel occlusions and stenoses, and aneurysm formation. Venous involvement typically includes superficial thrombophlebitis or deep venous thrombosis, usually of the lower extremities. Vena cava thrombosis can also occur, with extension to the hepatic vein, leading to Budd-Chiari syndrome and its associated morbidity and mortality. Patients with arterial manifestations may present with thrombosis or aneurysm formation with possible fatal rupture, especially if pulmonary arteries are involved.
-
GI: GI disease is associated with pyoderma gangrenosum and with papulopustular lesions, especially in children. [6] GI involvement consists of small vessel disease that involves mucosa and results in ulcers or large vessel disease that results in ischemia and infarction. The ileocecal region is the area most commonly involved with ulcers; the ulcers may be superficial or more typically deep with increased risk of perforation.
Race
Behçet syndrome is thought to be more common in Turkish, Asian, and Middle Eastern populations. However, the severity of disease may be increased in these populations, with better case ascertainment as a result. An increased incidence of skin pathergy and HLA-B51 antigen is observed in Middle Eastern and Asian patients, compared with North American or northern European patients.
Sex
In Japan and Korea, Behçet syndrome is more common in females, with a male-to-female ratio of 1:2, but it is more common in males in the Middle East, with a male-to-female ratio of 2:1. In the literature, estimates of male-to-female ratios range from 11:1 to 0.2:1. Despite the variability of the reported sex ratios, the disease tends to run a more severe course in males.
Age
Onset typically occurs in patients in the late third and early fourth decades of life. Onset during the childhood years is well recognized, but Behçet syndrome rarely occurs before school age. Mean age of onset for pediatric patients in a large Turkish series was 11.7 years, and the mean age of onset for neurological Behçet syndrome was 13 years.
-
MRI, T2-weighted images of brainstem involvement with meningoencephalitis in an 11-year-old girl with neurologic Behçet syndrome.
-
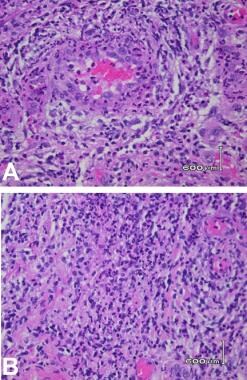
Histology of ulcers revealing neutrophilic infiltrate and vasculitis.






