Practice Essentials
Silver-Russell syndrome (SRS) originally was described by Silver and colleagues in 1953 and, soon afterwards, by Russell in 1954. [1, 2] Silver-Russell syndrome is characterized by intrauterine and postnatal growth retardation leading to a small-for-gestational-age (SGA) infant at birth, feeding difficulties during infancy, short stature, body asymmetry, characteristic triangular facies with prominent forehead, and several other anomalies.
Phenotypes in Silver-Russell syndrome range from mild to classic. Most patients are found to have hypomethylation in the chromosome 11p15 imprinting center 1 (IC1); some patients have maternal uniparental disomy of chromosome 7, with the possibility of imprinting (eg, inheriting two copies of maternal chromosome 7, with no paternal contribution). [3, 4, 5, 6]
Signs and symptoms of Silver-Russell syndrome
Dysmorphic facies
These include the following:
-
Classic features include normal head circumference (head may appear large because of small body size), blue sclerae, small triangular facies, a high forehead that tapers to a small jaw, micrognathia, prominent nasal bridge, and down-turning corners of the mouth
-
Mild dysmorphic facies include some features of the classic facies
Growth and skeletal
These include the following:
-
Prenatal onset short stature (final height ≤ -3.6 standard deviations [SD])
-
Late closure of anterior fontanelle
-
Asymmetry, usually of the limbs
-
Hemihypertrophy
-
Clinodactyly of the fifth finger
-
Camptodactyly
-
Syndactyly of second and third toes
-
Sprengel deformity or other hand anomalies [7]
Genital anomalies
These include the following:
Radiographic findings, hand
These include the following:
-
Delayed bone age
-
Ivory epiphyses of the distal phalanges
-
Small middle phalanx of the fifth finger (80%)
-
Pseudoepiphyses at the base of the second metacarpal
Other (occasional)
These include the following:
-
Cardiac defects
Workup in Silver-Russell syndrome
Silver-Russell syndrome remains a clinical diagnosis based on a combination of characteristic features and a scoring system.
Several scoring systems exist in the literature, but a published international consensus statement on the clinical diagnosis, investigation, and management of Silver-Russell syndrome endorsed the Netchine-Harbison clinical scoring system (NH-CSS), based on favorable sensitivity (98%) and negative predictive (89%) values. [8]
Laboratory studies
An NH-CSS–based diagnostic flow chart published in a 2017 international consensus statement on the diagnosis and management of Silver-Russell syndrome recommended 11p15 loss of methylation (LOM) and UPD(7)mat as the initial molecular studies in likely Silver-Russell syndrome.
Imaging studies
Plain film hand radiographic findings include those indicated above.
Management of Silver-Russell syndrome
A medical home with a multidisciplinary care facility is ideal for patients with Silver-Russell syndrome. Usually, a team may include the following:
-
Clinical genetic counselors
-
Dietitians and gastroenterologists - For feeding issues and failure to thrive
-
Endocrinologists - For short stature and puberty
-
Speech therapists - For delay
-
Orthopedists - For scoliosis
However, the team can be tailored according to the patient's specific symptoms and needs.
Growth monitoring
Careful monitoring of linear growth after optimizing caloric intake is critical for early diagnosis of catch-up growth failure by 2 years of age and early intervention with recombinant human growth hormone (rhGH) for maximizing final adult height.
Development
An early intervention program, including physical therapy, is beneficial. Special education courses are needed when the child is older.
Surgical care
Consider enteral feeding if the patient does not tolerate oral feeding and has severe failure to thrive. Nasogastric or percutaneous endoscopic gastrostomy (PEG) feeds are needed to facilitate growth and maintenance.
Pathophysiology
Growth failure is the primary abnormality. The American College of Radiology has established guidelines regarding growth disturbances. [9]
Patients typically present with intrauterine growth retardation, difficulty feeding, failure to thrive, or postnatal growth retardation. Adequate catch-up growth often does not occur, and final adult height still is less than normal (≤ -3.6 standard deviations [SD]). See the image below.
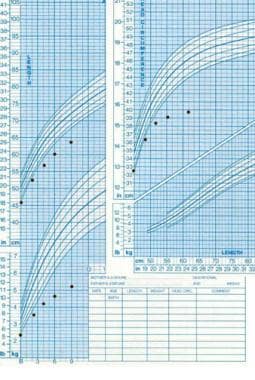 Failure of growth in weight, length, and head circumference starting at birth, suggesting an organic etiology that occurred in utero.
Failure of growth in weight, length, and head circumference starting at birth, suggesting an organic etiology that occurred in utero.
Older children and adults do not manifest clinical features as clearly as infants or young children. Growth hormone insufficiency may be present. Abnormalities of spontaneous growth hormone (GH) secretion and subnormal responses to provocative growth hormone stimulation testing have been reported in a significant number of children with Silver-Russell syndrome. Facial dysmorphism is observed, with small triangular facies and normal head circumference. Because length usually is less than normal, the head appears disproportionately large. Intelligence may be normal, or the patient may have a learning disability. The limbs may be asymmetrical, and camptodactyly (ie, fixed flexion of digits) or clinodactyly (ie, incurving) of one or more fingers may be present.
An observational study by Lokulo-Sodipe et al of adults with Silver-Russell syndrome indicated that persons with this condition have a higher fat percentage (median 44.4%), a higher fat mass index (median 9.6 kg/m2), and a higher trunk-to-limb fat ratio (median 1.2; demonstrating greater central adiposity) than do age- and sex-matched individuals without the syndrome (32.1%, 8.3 kg/m2, 0.88, respectively). Meanwhile, in the same two cohorts, total lean mass (median 30.8 kg) and total lean percentage (median 51.8%) were lower in persons with Silver-Russell syndrome than in the controls (48.1 kg, 65.1%, respectively). The investigators also found hypercholesterolemia, hypertriglyceridemia, and dysglycemia in the syndrome group, with 52.0% of the disease cohort having a total cholesterol level of 5 mmol/L or more and 20.8% having a triglyceride level of 1.7 mmol/L. In addition, 25.0% had diabetes mellitus or impaired fasting glycemia. With regard to metabolic syndrome, although the investigators state that two previous studies did not find metabolic syndrome in patients with Silver-Russell syndrome, this report, using different criteria than the other studies, detected metabolic syndrome in 18.2% of the disease cohort. [10]
Epidemiology
Frequency
International
Estimates of incidence range from as high as 1 case in 3000 population to as low as 1 case in 100,000 population.
Mortality/Morbidity
Infants have failure to thrive, feeding difficulties, and fasting hypoglycemia. A retrospective study by Marsaud et al found digestive problems and malnutrition to be common in children with Silver-Russell syndrome. The study, in which 75 patients (median age 24.3 mo) were assessed, found malnutrition in 70% of them and gastrointestinal signs in 77% of them. The latter included feeding difficulties (65%), severe gastroesophageal reflux (55%), severe vomiting before age 1 year (50%), persistent vomiting from age 1 year (29%), and constipation (20%). [11]
Sex
The male-to-female ratio is equal.
Age
Clinical features are easier to identify in infants and younger children, particularly the small triangular facies. These findings are more difficult to recognize in adults.
-
Failure of growth in weight, length, and head circumference starting at birth, suggesting an organic etiology that occurred in utero.






