Practice Essentials
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children (see the image below). In patients with localized disease, overall 5-year survival rates have improved to more than 80% with the combined use of surgery, radiation therapy, and chemotherapy. [1] However, in patients with metastatic disease, little progress has been made in survival rates, with a 5-year, event-free survival rate of less than 30%.
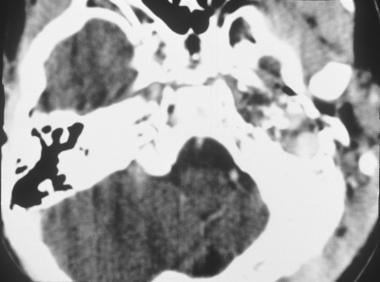 Axial CT scan of rhabdomyosarcoma in the left middle ear. Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals.
Axial CT scan of rhabdomyosarcoma in the left middle ear. Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals.
Signs and symptoms
Rhabdomyosarcoma usually manifests as an expanding mass. Tumors in superficial locations may be palpable and detected relatively early, but those in deep locations (eg, retroperitoneum) may grow large before causing symptoms.
Symptoms depend on the location of the tumor, and pain may be present. Typical presentations of nonmetastatic disease, by location, are as follows:
-
Orbit: Proptosis or dysconjugate gaze [2]
-
Paratesticular: Painless scrotal mass
-
Prostate: Bladder or bowel difficulties
-
Uterus, cervix, bladder: Menorrhagia or metrorrhagia
-
Vagina: Protruding polypoid mass (botryoid, meaning a grapelike cluster)
-
Extremity: Painless mass
-
Parameningeal (ear, mastoid, nasal cavity, paranasal sinuses, infratemporal fossa, pterygopalatine fossa): Upper respiratory symptoms or pain [3]
Metastatic disease may cause the following symptoms:
-
Bone pain
-
Respiratory difficulty (secondary to lung nodules or to pleural effusion)
-
Anemia
-
Thrombocytopenia
-
Neutropenia
Disseminated rhabdomyoblasts in the bone marrow may mimic the symptoms and light microscopic findings of leukemia.
See Clinical Presentation for more detail.
Diagnosis
Laboratory studies
-
Complete blood count (CBC): Anemia may be present because of inflammation, or pancytopenia may be present from bone marrow involvement
-
Liver function tests: Including lactic acid dehydrogenase (LDH), aspartate aminotransferase (AST), alanine aminotransferase (ALT), alkaline phosphatase, and bilirubin levels; metastatic disease of the liver may affect values of these proteins
-
Renal function tests: Including blood urea nitrogen (BUN) and creatinine levels
-
Urinalysis : Hematuria may indicate involvement of the genitourinary tract
-
Blood electrolyte and chemistry: Including sodium, potassium, chlorine, carbon dioxide, calcium, phosphorous, and albumin values
Liver and renal function, as well as blood electrolytes and chemistry, must be assessed before chemotherapy.
Genetic studies
-
Fluorescent in situ hybridization (FISH)
-
Reverse transcriptase–polymerase chain reaction (RT-PCR) assay: When FISH is unavailable or uninformative
Imaging studies
-
Plain radiography
-
Computed tomography (CT) scanning
-
Magnetic resonance imaging (MRI)
-
Bone scanning
-
Ultrasonography
-
Echocardiography
Biopsy
-
Open or core needle biopsy: To obtain tissue sampling for diagnosis and molecular studies
-
Bone marrow aspiration and biopsy: To assess for metastatic spread to bone marrow
See Workup for more detail.
Management
Treatment for patients with rhabdomyosarcoma involves a combination of surgery, chemotherapy, and radiation therapy.
Primary tumor
If possible, complete excision of the lesion should be performed with a wide (2-cm) margin of healthy tissue (although wide margins of normal tissue often are impossible to achieve at certain sites, such as the head and neck).
Most patients with rhabdomyosarcoma require radiation therapy to achieve adequate local control, though this treatment is not usually performed until after initial surgical resection and the start of chemotherapy.
Data from Europe suggest that chemotherapy alone can be effective for achieving adequate local control in some patients who have complete response of the primary tumor. However, surgery and/or irradiation are needed for local control of residual disease.
Lymph nodes
Regional lymph nodes that appear to be clinically or radiographically involved should be sampled to determine the tumor’s surgicopathologic clinical group and the need for later radiation therapy.
See Treatment or Medication for more detail.
Prevention
No preventive measures are known for childhood cancers.
Background
Rhabdomyosarcoma (RMS) is the most common soft tissue sarcoma in children, accounting for 4.5% of all cases of childhood cancer. It is the third most common extracranial solid tumor of childhood. [4, 5] RMS is a malignant tumor of mesenchymal origin that is included in the group of small round blue cell tumors of childhood along with neuroblastoma, lymphoma and primitive neuroectodermal tumors. The name is derived from the Greek words rhabdo, which means rod shape, and myo, which means muscle. Although Weber first described rhabdomyosarcoma in 1854, a clear histologic definition was not available until 1946, when Stout recognized the distinct morphology of rhabdomyoblasts. [6] Stout described rhabdomyoblasts as appearing in round, strap, racquet, and spider forms. As its name suggests, the tumor is believed to arise from a primitive muscle cell. Rhabdomyoblasts sometimes have discernible muscle striations that are visible on specimens under light microscopy, although electron microscopy may be needed to detect subcellular elements. Cells are usually positive for intermediate filaments and other proteins typical of differentiated muscle cells, such as desmin, vimentin, myoglobin, actin, and transcription factor myoD.
An image depicting rhabdomyosarcoma can be seen below.
Axial CT scan of rhabdomyosarcoma in the left middle ear. Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals.
RMS is stratified into different histologic subtypes which influence management plans and patient outcome. [7]
Several distinct histologic groups have prognostic significance, including embryonal rhabdomyosarcoma (ERMS), which occurs in 55% of patients; the botryoid variant of ERMS, which occurs in 5% of patients; alveolar rhabdomyosarcoma (ARMS), which occurs in 20% of patients; and undifferentiated sarcoma (UDS), which occurs in 20% of patients. [8] Similar to other pediatric malignancies, genomic sequencing studies show that RMS harbors a low DNA mutational burden.
Treatment responses and prognoses widely vary depending on location and histology. Studies of tumor biology and treatment in patients with rhabdomyosarcoma at a single institution, or even at regional centers, are not very feasible because of the variable nature and uncommon occurrence of the tumors. Therefore, most advances in knowledge and treatment have resulted from cooperative group studies.
Pathophysiology
The tumor is believed to arise from primitive muscle cells, but tumors can occur anywhere in the body; however, a primary bone rhabdomyosarcoma has not been reported. The most common sites are the head and neck (28%), extremities (24%), and genitourinary (GU) tract (18%). Other notable sites include the trunk (11%), orbit (7%), and retroperitoneum (6%). Rhabdomyosarcoma occurs at other sites in less than 3% of patients. The botryoid variant of ERMS arises in mucosal cavities, such as the bladder, vagina, nasopharynx, and middle ear. Lesions in the extremities are most likely to have an alveolar type of histology. Metastases are found predominantly in the lungs, bone marrow, bones, lymph nodes, breasts, and brain.
As with most tumors of childhood, the cause of rhabdomyosarcoma is unknown. The alveolar variant is so named because of the thin criss-crossing fibrous bands that appear as spaces between cellular regions of the tumor (reminiscent of lung alveoli). This variant is usually associated with 1 of 2 chromosomal translocations, namely, t(2;13) or t(1;13). These result in the fusion of the DNA-binding domain of the neuromuscular developmental transcription factors, encoded by PAX3 on chromosome 2 or PAX7 on chromosome 1 [9] , to the transcriptional activation domain of a relatively ubiquitous transcription factor, FKHR (or FOXO1a), which is encoded on chromosome 13. Less common translocations involving the PAX genes have been found in some rare cases. [10]
The resulting hybrid molecule is a potent transcription activator. It is believed to contribute to the cancerous phenotype by abnormally activating or repressing other genes. The embryonal subtype usually has a loss of heterozygosity at band 11p15.5; this observation suggests the presence of a tumor suppressor gene. Other molecular aberrations that may provide clues to the origin of the tumor and that may be useful for future treatment strategies include TP53 mutations (which occur in approximately one half of patients), an elevated N-myc level (in 10% of patients with ARMS), and point mutations in N-ras and K-ras oncogenes (usually embryonal). In addition, levels of insulinlike growth factor-2 may be elevated, suggesting pathways involving autocrine and paracrine growth factors. [11]
Etiology
The cause of rhabdomyosarcoma is unclear. Several genetic syndromes and environmental factors are associated with increased prevalence of rhabdomyosarcoma. [12]
-
Genetic syndromes include the following:
Neurofibromatosis (4-5% risk of any one of numerous malignancies)
Li-Fraumeni syndrome (germline mutation of the tumor suppressor gene TP53)
Costello syndrome [13]
-
A higher prevalence of congenital anomalies are observed in patients who later develop rhabdomyosarcoma with locations as follows:
Genitourinary (GU) tract
CNS (ie, Arnold-Chiari malformation)
GI tract
Cardiovascular system
-
Environmental factors appear to influence the development of rhabdomyosarcoma, as follows:
Parental use of marijuana and cocaine
Intrauterine exposure to X-rays [14]
Previous exposure to alkylating agents
Epidemiology
United States statistics
The incidence is 6 cases per 1,000,000 population per year (approximately 250 cases) in children and adolescents younger than 15 years.
International statistics
No notable geographic predilection is reported.
Race-, sex-, and age-related demographics
No racial predilection is obvious.
Overall, the male-to-female ratio is 1.2-1.4:1. Differences are observed according to the site of primary disease.
-
GU tract: The male-to-female ratio is 3.3:1 in patients with bladder or prostate involvement and 2.1:1 in rhabdomyosarcoma of the GU tract without bladder or prostate involvement.
-
Extremity: The male-to-female ratio is 0.79:1.
-
Orbit: The male-to-female ratio is 0.88:1.
Approximately 87% of patients are younger than 15 years, and 13% of patients are aged 15-21 years. Rhabdomyosarcoma rarely affects adults. Age-related differences are observed for the different sites of primary disease. Two age peaks tend to be associated with different locations. Patients aged 2-6 years tend to have head and neck or GU tract primary tumors, whereas adolescents aged 14-18 years tend to have primary tumors in extremity, truncal, or paratesticular locations.
-
GU tract: In patients with bladder or prostate involvement, 73% are younger than 5 years. In patients with rhabdomyosarcoma of the GU tract without bladder or prostate involvement, 27% are older than 15 years.
-
Orbit: About 42% of patients with orbital rhabdomyosarcoma are aged 5-9 years.
Prognosis
Morbidity/mortality
In patients with localized disease, overall 5-year survival rates have improved to more than 80% with the combined use of surgery, radiation therapy, and chemotherapy. [1] However, in patients with metastatic disease, little progress has been made in survival rates, with a 5-year event-free survival rate less than 30%. Those patients with metastatic disease without other high-risk factors, including unfavorable site, more than 3 sites, bone marrow involvement, and age younger than 1 year or older than 10 years, have a better prognosis (50% 3-y event-free survival) than those with 3-4 of these factors (12% and 5% 3-y event-free survival, respectively). [15] The use of high-dose myeloablative therapy with autologous stem-cell rescue has not improved outcomes for these patients. [16]
In an analysis of data collected by the Surveillance, Epidemiology, and End Results (SEER) program, mortality was highly related to age, site, and histology. [17] The 5-year survival was highest in children aged 1-4 years (77%) and was worst in infants and adolescents (47% and 48%, respectively). Orbital and GU sites were the most favorable (86% and 80%, respectively). Unfavorable sites included tumors of the extremities (50%), retroperitoneum (52%), and trunk (52%). Embryonal histology was best (67%) compared with alveolar histology (49%). Most patients with local recurrence are curable with salvage therapy, particularly if the recurrence is after initial therapy has been completed.
Complications
The treatment of rhabdomyosarcoma results in a multitude of potential long-term adverse effects. [18] The most common findings include the following:
-
Cardiomyopathy
In patients who receive an anthracycline, cardiac function must be monitored to assess for the development of cardiomyopathy.
Cardiomyopathy may also from cyclophosphamide use.
-
Pulmonary failure
-
Metabolic derangements: Ifosfamide use, in particular, can lead to renal electrolyte wasting (Fanconi syndrome).
-
Secondary malignant neoplasms
Secondary malignant neoplasms may arise as a result of radiotherapy and chemotherapy, particularly with alkylating agents.
Etoposide markedly increases the risk for acute myelogenous leukemia, particularly when regimens with frequent dosing schedules are used.
Radiation therapy increases the risk of second malignancies, including skin and bone tumors.
Patient Education
Chemotherapy
Parents and patients (if appropriate) must undergo formal training to learn about the adverse effects of chemotherapy. They must know what is expected to happen as a result of the therapy and are encouraged to ask questions.
Central venous catheters
When patients have central venous catheters that exit the skin (eg, Hickman or Broviac catheters), the parents or the patient must learn to properly care for the line. This care usually involves daily heparin flushes.
Patients and parents must understand the limitations on activities because of central venous catheters. For example, swimming is not permitted.
Patients with subcutaneous catheters (eg, Mediport catheters) do not need to perform daily care, but they should learn to apply a topical anesthetic (eg, EMLA cream, or lidocaine-prilocaine cream) at least 1 hour before an anticipated needle stick.
-
Axial CT scan of rhabdomyosarcoma in the left middle ear. Image provided by Suresh Muhkerji, MD, Department of Radiology, University of North Carolina Hospitals.






