Practice Essentials
Acanthocytosis is a red cell phenotype associated with various underlying conditions. Acanthocytes (from the Greek word acantha, which means thorn), or spur cells, are spiculated red cells with a few projections of varying size and surface distribution (see the images below). Studies in acanthocytosis workup include a complete blood count (CBC) and a peripheral blood smear. Management of acanthocytosis depends on the underlying condition. [1]
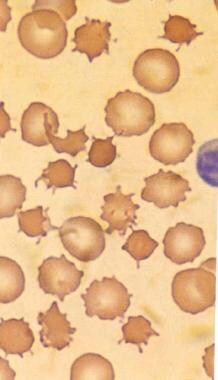 This image (magnified X 2000) shows the spiculated thorny RBCs (acanthocytes) as observed in an individual with abetalipoproteinemia. These are indistinguishable from the acanthocytes shown in the next image, which are observed in an individual with spur cell hemolytic anemia. Used with permission from Little, Brown and Company.
This image (magnified X 2000) shows the spiculated thorny RBCs (acanthocytes) as observed in an individual with abetalipoproteinemia. These are indistinguishable from the acanthocytes shown in the next image, which are observed in an individual with spur cell hemolytic anemia. Used with permission from Little, Brown and Company.
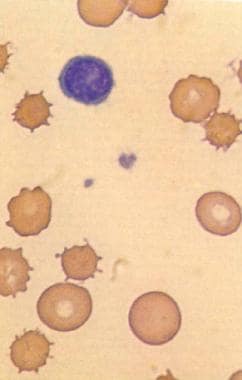 This image (magnified X 2000) demonstrates acanthocytes in an individual with spur cell hemolytic anemia associated with alcoholic cirrhosis. Acanthocytes, unlike echinocytes or burr cells, have fewer spicules. Used with permission from Little, Brown and Company.
This image (magnified X 2000) demonstrates acanthocytes in an individual with spur cell hemolytic anemia associated with alcoholic cirrhosis. Acanthocytes, unlike echinocytes or burr cells, have fewer spicules. Used with permission from Little, Brown and Company.
The cells appear contracted, dense, and irregular. The morphology of acanthocytes in these various conditions is similar, but the pathogenesis and clinical context often greatly differ. In general, the formation of acanthocytes depends on the alteration of the lipid composition and fluidity of the red cell membrane. [2]
The most frequent and most significant conditions underlying acanthocytosis include the following:
-
Abetalipoproteinemia - Bassen-Kornzweig syndrome if neurologic symptoms [3]
-
Spur cell hemolytic anemia of severe liver disease [4]
Other, less frequent conditions include the following:
-
Anorexia nervosa and other malnutrition states
-
Infantile pyknocytosis
-
In(Lu) null Lutheran phenotype
-
Idiopathic neonatal hepatitis
-
Myxedema
-
Transient hemolysis and stomatocytosis in individuals with alcoholism and mild hemolysis and spherocytosis in individuals with congestive splenomegaly
-
Homozygous familial hypobetalipoproteinemia
-
Zieve syndrome
-
Chronic granulomatous disease (CGD) associated with McLeod red cell phenotype
Acanthocytes should be distinguished from echinocytes (from the Greek word echinos, which means urchin). Echinocytes, or burr cells, appear with multiple small projections that are uniformly distributed on the red cell surface (see the image below). [10]
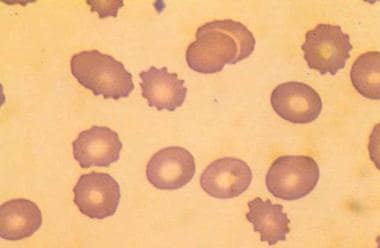 This image (magnified X 2000) shows echinocytes, or burr cells, a universal feature of uremia. The spicules of acanthocytes vary in length and width and project nonuniformly from the cell surface, while burr cells have regularly spaced, smoothly rounded crenulations. The second morphologic feature of RBCs in an individual with uremia is the presence of ellipsoid cells. Used with permission from Little, Brown and Company.
This image (magnified X 2000) shows echinocytes, or burr cells, a universal feature of uremia. The spicules of acanthocytes vary in length and width and project nonuniformly from the cell surface, while burr cells have regularly spaced, smoothly rounded crenulations. The second morphologic feature of RBCs in an individual with uremia is the presence of ellipsoid cells. Used with permission from Little, Brown and Company.
Echinocytes occur in many conditions, including malnutrition associated with mild hemolysis due to hypomagnesemia and hypophosphatemia, uremia, hemolytic anemia in long-distance runners, and pyruvate kinase deficiency. In vitro, elevated pH, blood storage, adenosine triphosphate (ATP) depletion, calcium accumulation, and contact with glass can lead to the formation of echinocytes.
Signs and symptoms of acanthocytosis
Signs and symptoms depend on the cause. Patients with acanthocytosis associated with abetalipoproteinemia may have a history of chronic diarrhea with pale, foul-smelling, and bulky stools; loss of appetite and vomiting; and slow weight gain and decreased growth, possibly with a bleeding tendency.
Patients with neuroacanthocytosis may report symptoms of ataxia, tremors, and visual abnormalities. Adolescents and adults may report dyskinesias, specifically orolingual and cognitive deterioration.
Patients with hemolysis may have jaundice, abdominal pain, pallor, dark urine, and recurrent infections.
Workup in acanthocytosis disorders
A complete blood count (CBC) reveals mild to moderate normocytic anemia with an elevated reticulocyte count. Peripheral blood smear findings reveal 0.2-90% acanthocytes.
The plasma lipid profile may be abnormal. In abetalipoproteinemia, plasma cholesterol levels are very low, less than 50 mg/dL. Plasma phospholipid levels are very low, while plasma apolipoprotein B, chylomicrons, very–low-density lipoproteins (VLDLs), and low-density lipoproteins (LDLs) are absent. Serum triglyceride levels are very low, less than 10 mg/dL. Plasma sphingomyelin levels are relatively increased at the expense of lecithin. [11]
Management of acanthocytosis disorders
Treatment of disorders with acanthocytosis depends on the underlying condition. Medical care in abetalipoproteinemia includes dietary restriction of long-chain fatty acids, with judicious supplementation with medium-chain triglycerides. Supplementation with lipid-soluble vitamins A, D, E, and K is necessary in large doses.
Typical care for severe liver disease includes careful fluid management, correction of metabolic disturbances, treatment of hypoglycemia, and careful nutritional management.
Pathophysiology
The description of acanthocyte pathophysiology varies depending on the underlying condition. Acanthocytes can be caused by (1) altered distribution or proportions of membrane lipids or by (2) membrane protein or membrane skeleton abnormalities. In membrane lipid abnormalities, previously normal red cell precursors often acquire the acanthocytic morphology from the plasma. Altered membranes may contain decreased phosphatidylcholine levels but increased levels of cholesterol and sphingomyelin. The imbalance in membrane lipids causes cells to stiffen, wrinkle, pucker, and form spicules because of a relative increase of the outer hemileaflet's surface area compared with the inner hemileaflet's surface area. In membrane protein or membrane skeleton abnormalities, the defect is intrinsic but, again, causes imbalances in inner versus outer leaflet surface areas and abnormal interaction between the membrane skeleton and lipid membrane.
The alteration in plasma lipids in autosomal recessive abetalipoproteinemia caused by the absence of beta-apolipoprotein is best described. Specifically, the lipoproteins apoprotein B (ApoB)–48 and ApoB-100 are deficient because of either abnormal assembly or defective aposecretion, leading to absent cellular secretion from hepatocytes or intestinal epithelial cells. Formation of normal chylomicrons (ie, lipoproteins that contain cholesterol and triglycerides) is inhibited and prevents intestinal absorption of lipids, leading to severe fat malabsorption. In addition to lipid abnormalities and altered membrane integrity, red cells have secondary vitamin E–deficiency and develop increased oxidant sensitivity, with a tendency to hemolyze more easily. Acanthocytes in the homozygous form of familial hypobetalipoproteinemia are thought to have a similar pathophysiology.
Severe liver dysfunction of various etiologies can also cause altered plasma lipid composition and acanthocytes (usually called spur cells in this case) because of acquired abnormal red cell membrane lipid composition. The liver dysfunction causes the accumulation of an abnormal, apolipoprotein A-II-deficient lipoprotein in plasma. Red cells are loaded with cholesterol by this lipoprotein and acquire an increased cholesterol-to-phospholipid ratio and a surface area preferentially within the outer bilayer leaflet. Cholesterol-laden red cells are then remodeled in the spleen, resulting in the typical spur cell shape. The molecular mechanisms are unclear but may include influences on membrane protein content and functions. The resulting spur cells are less deformable and are easily trapped in the spleen, conferring markedly shortened red cell survival.
Another group of patients with liver dysfunction may have normal red cell membrane lipids, and the pathophysiology in these cases is unknown. This situation is seen more often in children with severe hepatocellular dysfunction.
In neuroacanthocytosis, the plasma lipoproteins are normal and the formation of acanthocytes may be associated with intrinsic membrane abnormalities. Studies have indicated that abnormal protein formation or membrane protein trafficking is involved. Electron microscopy studies have shown that membrane protrusions depend on the irregular distribution of the membrane skeletons. The pathophysiology of the neurologic findings may also relate to the abnormal protein distribution, and in chorea-acanthocytosis syndrome, there are abnormal depositions of several sphingolipids and phospholipids in the brain. [12]
Abnormal lipid arrangements were also found in acanthocytes resulting from the McLeod blood group. Individuals with the McLeod blood group or McLeod syndrome lack the Kx antigen, a membrane precursor of the Kell antigen, which leads to acanthocytic red cell morphology. The Kell antigen is located on a 93-kD glycoprotein and is associated with the underlying membrane skeleton. Significant ultrastructural variability between cells is observed. McLeod acanthocytes also exhibit increased mechanical stability on ektacytometry findings, increased membrane rigidity, decreased potassium content, and an increased frequency of dense cells. Another blood group phenotype, the null Lutheran blood group or In(Lu) Lu(a-b-) red cell phenotype also causes acanthocytes. However, in both blood groups and in neuroacanthocytosis, cells are more resistant to hemolysis than in the previously mentioned disorders.
Acanthocytes are also found in myxedema, in panhypopituitarism, and in 20-65% of hypothyroidism cases. Serum lipid abnormalities with hypothyroidism are common, and patients with acanthocytes may have more severely abnormal lipids than patients with normal-shaped red cells.
Epidemiology
Frequency
International
Acanthocytes are found in 50-90% of cells on peripheral blood smear findings in abetalipoproteinemia, which is a rare autosomal recessive disorder with only about 100 cases described worldwide. Acanthocytes are also relatively common in severe liver dysfunction and malnutrition. Spur cell hemolytic anemia of severe liver disease is an uncommon complication and depends on the incidence of the underlying hepatic or hepatotoxic disorder. It occurs most often in patients with alcoholic cirrhosis, which develops in 10-30% of all patients with alcoholism (approximately 10 million in the United States).
The percentage of acanthocytes is usually smaller in the rare conditions of neuroacanthocytosis and McLeod and null Lutheran red cell blood group abnormalities. In hypothyroidism, 20-65% of cases have approximately 0.5-2% acanthocytes on peripheral smear findings.
Mortality/Morbidity
Mortality and morbidity with acanthocytosis due to abetalipoproteinemia is not well described because of the rarity of the disease and the limited prognostic data. Lifespan may be near normal with early diagnosis and adequate vitamin supplementation and dietary restriction but may vary significantly. Death may occur in the second or third decade and is usually determined by the degree and progression of neurological complications. Acanthocytosis due to severe liver dysfunction is a hallmark of high risk for mortality. In neonatal hepatitis, the process resolves in 65% of individuals within weeks to months. Acanthocytosis in infantile pyknocytosis is a transient benign process. In malnutrition, hypothyroidism, and myxedema, the red cell abnormality resolves with appropriate treatment and resolution of the underlying disease.
A prospective study by Alexopoulou et al indicated that in patients with liver cirrhosis, those with spur cell anemia have earlier mortality than those who do not. The study, which included 116 patients with cirrhosis, reported survival rates of 77%, 45%, and 33% in patients with spur cell anemia at 1-, 2-, and 3-month follow-up, respectively. [13]
Race
Acanthocytosis within the various underlying conditions is seen in all ethnicities.
Sex
Acanthocytosis has no sex predominance.
Age
The appearance of acanthocytes depends on the underlying condition. Acanthocytes in infants and children may indicate infantile pyknocytosis, neonatal hepatitis, autosomal recessive abetalipoproteinemia or homozygous familial hypobetalipoproteinemia, McLeod blood group, null Lutheran blood group, CGD with McLeod blood group, hypothyroidism, or severe malnutrition.
Medical/Legal Pitfalls
Acanthocytes should not be confused with echinocytes.
Abetalipoproteinemia is a rare disorder in children. The diagnosis may initially be missed or delayed.
Autosomal recessive abetalipoproteinemia must be differentiated from the homozygous form of familial hypobetalipoproteinemia. The presentation of both disorders is similar, but hypobetalipoproteinemia is clinically milder.
-
This image (magnified X 2000) shows the spiculated thorny RBCs (acanthocytes) as observed in an individual with abetalipoproteinemia. These are indistinguishable from the acanthocytes shown in the next image, which are observed in an individual with spur cell hemolytic anemia. Used with permission from Little, Brown and Company.
-
This image (magnified X 2000) demonstrates acanthocytes in an individual with spur cell hemolytic anemia associated with alcoholic cirrhosis. Acanthocytes, unlike echinocytes or burr cells, have fewer spicules. Used with permission from Little, Brown and Company.
-
This image (magnified X 2000) shows echinocytes, or burr cells, a universal feature of uremia. The spicules of acanthocytes vary in length and width and project nonuniformly from the cell surface, while burr cells have regularly spaced, smoothly rounded crenulations. The second morphologic feature of RBCs in an individual with uremia is the presence of ellipsoid cells. Used with permission from Little, Brown and Company.







