Practice Essentials
5-Alpha-reductase type 2 deficiency (5-ARD) is an autosomal recessive sex-limited condition resulting in the inability to convert testosterone to the more physiologically active dihydrotestosterone (DHT). Because DHT is required for the normal masculinization of the external genitalia in utero, genetic males with 5-alpha-reductase type 2 deficiency are born with ambiguous genitalia (ie. 46,XY disorder of sexual development (DSD)). [1]
Patients with 5-alpha-reductase type 2 deficiency classically present with striking ambiguity of the genitalia, with a clitoral-like phallus, bifid scrotum, pseudovaginal perineoscrotal hypospadias, and a rudimentary prostate. Occasionally, patients can appear more masculinized; they may lack a separate vaginal opening, and have isolated penile hypospadias [2] or even a penile urethra. [3]
The uterus and fallopian tubes are absent because of the normal secretion of the müllerian-inhibiting factor. Testes are intact and are usually found in the inguinal canal or scrotum; however, cryptorchidism is frequently described with testes occasionally located in the abdomen. Wolffian duct differentiation is normal with seminal vesicles, vasa differentia, epididymides, and ejaculatory ducts. The prostate is small, nonpalpable, and rudimentary in adulthood. Neither benign prostate hyperplasia (BPH) nor prostate cancer has been reported in these patients. [4]
The cause of 5-alpha-reductase type 2 deficiency is the deficiency of the type 2 isozyme of 5-alpha-reductase. As with most single enzyme disorders, 5-alpha-reductase type 2 deficiency is an autosomal recessive trait and sex limited because the clinical syndrome only affects genetic males, with very subtle phenotypic findings in homozygous females. [5]
Pathophysiology
The root cause of this disorder is a deficiency in the 5-alpha-reductase type 2 isoenzyme, which transforms testosterone to DHT. DHT is a more potent androgen than testosterone and is bound selectively to the androgen receptors in genital skin and fibroblasts. The 5-alpha-reductase type 2 isoenzyme is expressed in external genital tissues early in gestation, making its action necessary for the development of normal male external genital anatomy in the fetus. [3] As with most single enzyme disorders, 5-alpha-reductase type 2 deficiency is autosomal recessive and sex limited because it only causes a clinically significant disorder in genetic males, with very subtle phenotypic changes in homozygous females. See the image below.
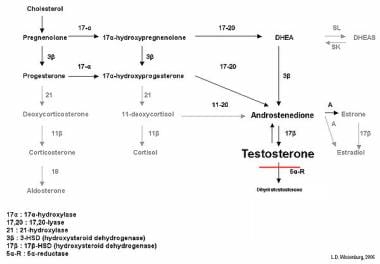 Biochemical effects of 5-alpha-reductase type 2 deficiency in testosterone biosynthesis. Typically levels of testosterone are elevated, whereas levels of dihydrotestosterone (DHT) are significantly decreased, leading to male undervirilization.
Biochemical effects of 5-alpha-reductase type 2 deficiency in testosterone biosynthesis. Typically levels of testosterone are elevated, whereas levels of dihydrotestosterone (DHT) are significantly decreased, leading to male undervirilization.
Three genes coding for 5-alpha-reductase have been identified, each for a slightly different isoenzyme. Only the genes coding for 5-alpha-reductase type 1 (SRD5A1) and 5-alpha-reductase type 2 (SRD5A2) are pertinent to the conversion of testosterone to DHT, whereas the gene coding 5-alpha-reductase type 3 is unrelated to any disorders of male sexual development, and may be implicated in a rare disorder of glycosylation. [6] Both of the pertinent genes, SRD5A1 and SRD5A2, contain five exons separated by four introns. A predominance of mutations have been observed in exons 1 and 4. [7, 8] The gene for 5-alpha-reductase type 2 has been determined to be on band 2p23. Lack of expression of this gene clinically correlates with the symptoms of 5-alpha-reductase type 2 deficiency. In adulthood, the 5-alpha-reductase type 2 isoenzyme is expressed in high levels in the prostate, genital skin, epididymis, seminal vesicle, and liver. [3]
The gene for 5-alpha-reductase type 1 is located on band 5p15. Its product is expressed only in nongenital skin and liver at low levels from the time the individual is aged 3 years until puberty, at which time enzyme expression is measurable in sebaceous glands and scalp. Linkage analysis has demonstrated that the type 1 enzyme is unrelated to the clinical syndrome of 5-alpha-reductase type 2 deficiency. Interestingly, partial virilization of males with 5-alpha-reductase type 2 deficiency occurs at puberty and may be attributable to the rise in type 1 enzyme activity or expression at that time. [4] An alternative mechanism for this virilization at puberty may be through binding of testosterone to the androgen receptor or by overcoming a partial enzyme deficiency with the increase in serum testosterone levels during puberty. [9]
More than 60 different mutations of the gene for 5-alpha-reductase type 2 have been reported in people with clinical and biochemical evidence of the enzyme deficiency. The occurrence of identical mutations in individuals with widely divergent geographic and ethnic backgrounds suggests the existence of mutational hot spots. [10] Patients with a heterozygous mutation have been shown to have a wider phenotypic spectrum; however, correlation between the severity of the syndrome and a particular gene defect has not been observed. [3] There is also a lack of phenotype/genotype relationship in patients carrying the same mutation, which suggests that factors other than 5-alpha-reductase enzyme activity contribute to phenotype. [11]
Batista and Mendonca found 129 different allelic variants in the SRD5A2 gene in the literature among persons with 5-alpha-reductase type 2 deficiency. The majority of the variants are missense mutations (n = 83); however, small deletions (n = 12), splicing mutations (n = 6), stop codons (n = 4), small indels (n = 20) and gross deletions (n = 4) have also been reported. [12]
Epidemiology
United States statistics
The carrier frequency and number of individuals with this disorder are not established.
International statistics
Although frequencies for various countries are not established, increased frequency is reported in the Dominican Republic, some highland tribes in New Guinea, and in Turkey. The high frequency in these areas represents the effect of consanguinity in specific kindreds. [3] Overall, more than 50 families with this disorder have been described in several parts of the world. In a few patients with 46,XY DSD due to 5-alpha-reductase type 2 deficiency diagnosed by clinical and hormonal findings, no mutations were identified in the SRD5A2. [13] In general, disorders of sexual differentiation as a whole are uncommon, with an overall incidence of 1:5500. [14]
Sex- and age-related demographics
Clinical 5-alpha-reductase type 2 deficiency is limited to genetic males. Although the enzyme deficiency can be documented in homozygous females, no clinical or developmental need for DHT is documented in women, who are likely asymptomatic.
Most individuals with 5-alpha-reductase type 2 deficiency are identified in the neonatal period with ambiguous genitalia. [15] However, some of these children are misdiagnosed as having partial or complete androgen insensitivity syndrome (AIS), which can produce almost identical phenotypes. As noted above, some patients with 5-alpha-reductase type 2 deficiency present with more masculinized genitalia including isolated hypospadias or penile urethra with micropenis, which can lead to a delay in diagnosis. [3] Delayed diagnosis can also occur in patients with isolated clitoromegaly and otherwise normal female-appearing external genitalia. In patients with this phenotype, diagnosis is often delayed until the time of puberty, when they present with primary amenorrhea and virilization. [10, 16] Preliminary data suggest the incidence of 5-alpha-reductase deficiency in elite female athletes that screen positive for hyperandrogenemia may be more than 200 times that of the general population. [17]
Medical/Legal Pitfalls
The clinical heterogeneity resulting in unpredictable and varying outcomes for patients with a DSD, particularly those associated with abnormalities in androgen response or synthesis, places an ethical, and potentially legal, burden on the treatment team. The team is responsible for ensuring education of the parent, and, when appropriate, the patient, regarding the diagnosis and obtaining informed consent prior to medical or surgical intervention. This is especially important in the setting of gonadectomy and/or genitoplasty, which are irreversible procedures that result in potential loss of fertility and have lifelong impact on gender satisfaction.
The healthcare team needs to clearly communicate to the parent of the patient and the patient, when appropriate, that available research is insufficient to absolutely predict adult gender satisfaction, irrespective of initial medical or surgical treatment. Early genitoplasty with or without gonadectomy does not appear to be associated with increased gender assignment satisfaction or a decrease in the potential for gender role change. On the other hand, although delaying these procedures provides a greater opportunity for the patient's involvement in the decision process, this alternative approach is not without risk. Some data suggest negative consequences of delayed repair of ambiguity on the development of self-esteem and gender satisfaction within cultures with low incidence of disease. [18, 19, 20]
The more contemporary approach, to delay definitive therapy, has developed over the last 2 decades as adult DSD patients and advocates have voiced their dissatisfaction with life-changing procedures performed prior to their ability to consent.
The consequence is that the parents of the newborn with a 46,XY DSD are placed in a difficult situation. They must adapt to the diagnosis, acutely acquire an understanding of the issues of gender as well as the risks, benefits, and potential complications of treatment and non-treatment options. This education process, ultimately concluded with a statement of informed consent, should satisfy a "reasonable patient" standard: "a doctor must disclose those risks, benefits, and alternatives that a reasonable person in the patient's position would be likely to consider significant in deciding whether or not to undergo treatment, including diagnosis and prognosis, common risks of the proposed procedure, remote risks with grave consequences, probable outcomes and expected posttreatment course, and the risks, benefits and unknowns of alternative treatments and non-treatment." [19]
Prognosis
Morbidity/mortality
This enzyme deficiency is not life threatening; however, if intra-abdominal testes are retained, an increased risk of gonadoblastoma is noted. Secondary issues include a risk of osteoporosis if hormone replacement therapy is not initiated in the patient with a gonadectomy. Psychological morbidity is not uncommon, with occasional asynchrony of assigned gender and sexual identity (see Treatment).
Complications
Psychosexual dysfunction may occur in the presence of genital malformation with or without surgical intervention. Appropriately timed consultations for patient and family to mental health professionals experienced in working with patients with DSD is important.
A mental health professional, preferably a child psychiatrist or pediatric psychologist, should be available to the patient and family due to the emotional loading of some of these issues. In addition to emotional support, they can help facilitate communication between the medical team, the patient, and the family.
Patient Education
Questions about gender and sexuality are extremely anxiety provoking and emotionally upsetting. Ensuring that the family feels informed and involved in the entire decision-making process is important. The family should leave the first session with a sense that all members of the team are in place to support them and should be provided the contact information that allows them to readily access any and all members of the treatment team.
Counseling is a lifelong process because of the variability of family dynamics, childhood development, and unforeseeable developments in medical and surgical treatment options. As the child grows, they must also feel a sense of comfort in accessing the team members to discuss age-appropriate developmental concerns to ultimately include intimacy with partners, sexual function and satisfaction, and potential fertility.
Education is a key part of the care plan for individuals with 5-alpha-reductase type 2 deficiency. Providing patients (and their families) with accurate, complete, and unbiased information about the diagnosis and the treatment options is an absolute requirement. This approach should include an honest discussion on the unpredictability of choosing gender identity and gender satisfaction.
Currently no group is specific to individuals with 5-alpha-reductase type 2 deficiency, but the androgen insensitivity syndrome (AIS) support group may be a reasonable surrogate. It has branches in several countries and maintains an active web site (see Androgen Insensitivity Syndrome Support Group: www.aissg.org). Members have made the group available to individuals with 46,XY DSD other than AIS.
Lastly, several Web sites may provide further information, including the following:
-
Biochemical effects of 5-alpha-reductase type 2 deficiency in testosterone biosynthesis. Typically levels of testosterone are elevated, whereas levels of dihydrotestosterone (DHT) are significantly decreased, leading to male undervirilization.
-
Prader scale reflecting the degree of virilization of the external genitalia. The internal genitalia reflect the changes in the urogenital sinus in response to the presence or absence of mullerian inhibiting hormone (MIH)









