Practice Essentials
Pyruvate kinase deficiency, one of the most common enzymatic defects of the erythrocyte, manifests clinically as a hemolytic anemia that can range from a mildly compensated anemia to severe anemia of childhood. It is caused by mutations in the PKLR gene. Surprisingly, however, the symptomatology is less severe than hematologic indices indicate. This is presumably due to enhanced oxygen delivery as a result of the defect. [1] [2, 3, 4, 5, 6, 7]
Most affected individuals do not require treatment, although in the most severe cases, death may occur in utero as a result of severe anemia. Periodic blood transfusions or splenectomy may be required, but most of the symptomatology is limited to early life and to times of physiologic stress or infection.
An international, multicenter registry that collected clinical data on patients with pyruvate kinase deficiency found that 93% of newborns were treated with phototherapy, and 46% were treated with exchange transfusions. Splenectomy was performed in 150 of 254 patients, or 59%, and was associated with a median increase in hemoglobin levels of 1.6 g/dL along with a decreased transfusion burden in 90% of patients. Predictors of a response to splenectomy included higher presplenectomy hemoglobin, lower indirect bilirubin, and missense PKLR mutations. In total, 87 of 254 patients, or 34%, had both a splenectomy and cholecystectomy. In patients who had a splenectomy without simultaneous cholecystectomy, 48% later required a cholecystectomy. [6]
(See the image below.)
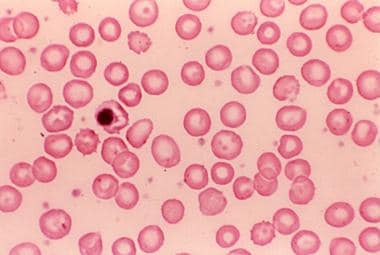 Peripheral blood smear in a child with splenectomy and pyruvate kinase deficiency.
Peripheral blood smear in a child with splenectomy and pyruvate kinase deficiency.
Signs and symptoms
The following are evident in pyruvate kinase deficiency:
-
Mild to severe normochromic and normocytic anemia
-
Reticulocytosis
-
Symmetrical growth delay
-
Failure to thrive
-
Cholecystolithiasis: Usually after the first decade of life but possibly in childhood
-
Frontal bossing
-
Hyperbilirubinemia in the newborn
-
Icteric sclera
-
Mild to moderate splenomegaly
-
Upper-right-quadrant tenderness
-
Murphy sign
-
Chronic leg ulcers (adults)
The birth history of patients with pyruvate kinase deficiency may include severe anemia, severe jaundice, [8] kernicterus, and a history of exchange transfusion.
Diagnosis
The minimal tests required to guide the investigation of pyruvate kinase deficiency include the following:
-
Complete blood count (CBC) with differential
-
Reticulocyte count
-
Serum bilirubin level study
-
Peripheral blood film examination
Normochromic, normocytic, or macrocytic anemia, together with reticulocytosis in the absence of blood loss, is suggestive of hemolysis. A negative Coombs test result helps to exclude immune hemolysis.
The enzyme activity rate in most patients with pyruvate kinase deficiency is 5-25% of normal, with measurement of the intermediates (2,3-diphosphoglycerol and glucose-6-phosphate) proximal to the enzyme defect helping to confirm the diagnosis.
Other findings include the following:
-
Normoblastic erythroid hyperplasia of the bone marrow
-
Extramedullary hematopoiesis
-
Splenic and hepatic hemosiderosis and splenic congestion
-
Reticuloendothelial hyperplasia
-
Erythrophagocytosis
-
Increased iron stores
Enzyme assay, as well as deoxyribonucleic acid (DNA) analysis with a polymerase chain reaction (PCR) assay or single-strand conformation polymorphism, can also be used to confirm the diagnosis of pyruvate kinase deficiency.
Management
In patients with mild to moderate pyruvate kinase deficiency, care is predominantly supportive. Red blood cell transfusion may be necessary if the patient’s hemoglobin value falls significantly.
Supplemental folic acid is used extensively in individuals with hemolytic anemia to prevent the development of megaloblastic anemia.
Splenectomy
Splenectomy is indicated only for patients with severe anemia. [9] The procedure does not abolish hemolysis or improve mild anemia, but it can reduce severe anemia and is frequently performed to minimize or eliminate the patient's need for blood transfusion.
Pharmacotherapy
Mitapivat is the first disease-modifying therapy approved for hemolytic anemia in adults with pyruvate kinase deficiency. It improves hemoglobin values and reduces transfusion burden in patients with pyruvate kinase deficiency by targeting the underlying defect. [10]
Pathophysiology
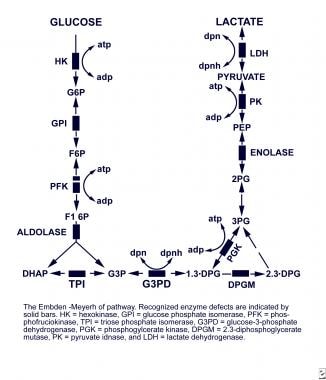
Embden-Meyerhof pathway
Erythrocytes (mature red blood cells) completely depend on glucose as a source of energy. Glucose is usually catabolized to pyruvate and lactate in the Embden-Meyerhof pathway, which is the major anaerobic glycolytic pathway. In the process, adenosine triphosphate (ATP), which is essential to providing the erythrocyte with energy, is generated. ATP plays a major role in maintaining a cation gradient in the erythrocyte, thus protecting the cell from premature death.
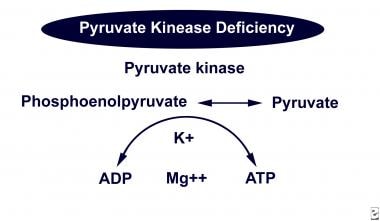
Pyruvate kinase catalyzes the conversion of phosphoenolpyruvate to pyruvate in the Embden-Meyerhof pathway, resulting in the production of ATP.
(See the images below.)
Erythrocytes in pyruvate kinase deficiency
In pyruvate kinase deficiency, an erythrocyte enzymopathy, a metabolic block is created in the Embden-Meyerhof pathway at the level of the deficient enzyme. Intermediate byproducts and various glycolytic metabolites proximal to the metabolic block accumulate in the erythrocyte, while the erythrocyte becomes depleted of the distal products in the pathway, such as lactate and ATP.
The lack of ATP disturbs the cation gradient across the erythrocytic cell membrane, causing the loss of potassium and water, which results in cell dehydration, contraction, and crenation (echinocytes) and leads to premature destruction of the erythrocyte.
Despite the resulting severe anemia, however, the high level of 2,3-diphosphoglycerate (2,3-DPG) increases the patient's exercise tolerance by causing a rightward shift in the hemoglobin-oxygen dissociation curve.
This is particularly advantageous during pregnancy because it enhances the transfer of oxygen to fetal blood, and it most likely adds to the particularly benign course of pyruvate kinase deficiency in many affected individuals. Women with the disorder typically do not require transfusions during pregnancy.
Pyruvate kinase–deficient reticulocytes can circumvent their defect by using the oxidative phosphorylation pathway to produce ATP. This ability is diminished when the reticulocytes are exposed to hypoxia or when they mature into erythrocytes. This may explain (1) the ineffective erythropoiesis in the spleen of patients with pyruvate kinase deficiency, (2) why most of the hemolysis occurs when the reticulocytes are trapped in the hypoxic environment of the spleen, and (3) the paradoxic increase in reticulocytes after splenectomy.
(See the image below.)
Peripheral blood smear in a child with splenectomy and pyruvate kinase deficiency.
Isoenzymes
Pyruvate kinase exists as 4 isoenzymes. Two isoenzymes (PKM1 and PKM2) are encoded by a genetic locus on band 15q22 and are found in striated muscle, the brain, fetal tissue, leukocytes, platelets, lungs, the spleen, the kidneys, and adipose tissue. The other 2 isoenzymes (PKL and PKR) are encoded by a genetic locus on band 1q21 and are found in the liver, normoblasts, reticulocytes, and erythrocytes. [11, 12, 13, 14] The liver and erythroid precursors are capable of activating PKM2 activity, but this is not the enzyme used under normal conditions. [15]
In persons with pyruvate kinase deficiency, band 1q21 is defective, resulting in deficient liver and red blood cell isoenzymes. The liver can compensate for the genetic defect in 2 ways. First, because the enzyme deficiency results in a less efficient enzyme rather than a nonfunctioning one, a greater quantity of enzyme can be produced. In addition, the liver can use residual PKM2 activity. Early in maturation, erythroid precursors use the PKM2 isoenzyme. As the cell matures, however, the PKR isoenzyme replaces the PKM2 enzyme. Because the erythrocyte cannot produce any new protein, it cannot compensate by increasing the quantity of isoenzyme or by using residual PKM2 isoenzyme.
Enzyme defects that have been described include decreased substrate affinity, increased product inhibition, decreased response to activator, and thermal instability. Mutations that strongly perturb enzyme kinetics and thermostability are associated with severe pyruvate kinase deficiency. [16] One severe form of the disease, pyruvate kinase Beppu, is associated with persistence of the PKM2 isoenzyme.
Iron overload
Iron overload is a serious, but not an unusual, complication in pyruvate kinase deficiency, even in patients not receiving chronic transfusion. These patients are not different from others with chronic hemolysis, who tend to absorb more iron regardless of their iron storage status because of the associated active erythropoiesis. Patients with iron overload not related to chronic hemolysis, such as in hemochromatosis, are usually protected from absorbing more iron.
The cause of such discrepancy was not clear until the discovery that a hepatic peptide known as hepcidin is a negative master regulator of iron absorption and release. In inflammation, the upregulated hepcidin prevents iron absorption, whereas, in iron deficiency anemia, a down-regulated hepcidin allows iron to be absorbed.
The loss of protection against iron absorption in patients with iron overload who have chronic hemolysis has been shown to be mediated by growth differentiation factor 15 (GDF15), a marrow factor that abrogates the effect of hepcidin. [17] In a study, the hepcidin level in patients with pyruvate kinase deficiency was 13-fold less than in the control group, whereas GDF15 was significantly higher in patients with pyruvate kinase than in control subjects.
Etiology
Acquired pyruvate kinase deficiency
Medical conditions such as acute leukemia, preleukemia, and refractory sideroblastic anemia, as well as complications from chemotherapy, can cause an acquired pyruvate kinase deficiency. This type is more common and milder than the hereditary type.
Hereditary pyruvate kinase deficiency
More than 180 genetic defects of the PK gene have been detected. [11, 12, 14, 18, 19] Most of these are missense mutations, but splicing mutations, insertions, and deletions also have been identified. Although inheritance is clinically autosomal recessive, most affected individuals are compound heterozygous for 2 different mutant alleles. [20] Homozygous individuals are usually the product of consanguineous mating. [14, 21, 22]
Heterozygotes have intermediate enzyme levels and are usually asymptomatic, while homozygotes manifest the clinical symptoms of pyruvate kinase deficiency.
The severity of the condition varies even among patients with the same level of deficiency because, in addition to the symptomatic homozygotes, compound heterozygotes with 2 different mutations (one can be qualitative and the other quantitative) also vary symptomatically.
Epidemiology
Occurrence in the United States
Pyruvate kinase deficiency and glucose-6-phosphate deficiency (G6PD) are the most common erythrocyte enzymopathies, with pyruvate kinase deficiency being the most common enzymopathy of anaerobic glycolysis. The prevalence rate of heterozygous carriers of 1 deficient PK gene is believed to be approximately 1%, although a population survey in Ann Arbor, Michigan, revealed a rate of 0.14%. Beutler and Gelbart estimated the incidence of PKD in the United States to be 1:20,000. It is known, however, that the incidence is much higher in remote and exclusive communities such as the Amish in Pennsylvania and Ohio and some communities in southern Utah. [23]
US population screening for the 4 most common gene mutations in pyruvate kinase deficiency demonstrated an estimated prevalence of 51 cases per million persons in the white population. [23] This is 50 times higher than the number of individuals who were diagnosed with the disease at a major pyruvate kinase assay laboratory in the United States over a 25-year period, suggesting that the deficiency is underdiagnosed.
As previously indicated, pyruvate kinase deficiency is particularly common among the Pennsylvania Amish, in whom the disorder can be traced to a single immigrant couple. Affected individuals can have severe, life-threatening manifestations and may require long-term transfusion therapy and splenectomy in early childhood. [24, 25]
International occurrence
Although pyruvate kinase deficiency occurs worldwide, most cases have been reported in northern Europe and Japan, as well as in the United States. In India, a study to screen newborns with jaundice for the presence of pyruvate kinase deficiency determined that 3.21% of all newborns with jaundice also had the deficiency, with a 30-40% reduction found in pyruvate kinase activity. [26]
Frequent reports of the predominance of pyruvate kinase deficiency in individuals of northern European ancestry can be questioned based on the increased number of cases that have been reported in different countries and in various non–northern European ethnic groups. Increased access to advanced medical facilities by these groups is assumed to be responsible for many of the reports, indicating that the prevalence of pyruvate kinase deficiency previously reported in persons of northern European ancestry matches that in persons of other ethnicities. [27]
The prevalence rate of heterozygous carriers with 1 deficient PK gene has been estimated to be 1% in Germany, 6% in Saudi Arabia, and 3% in Hong Kong.
Race-related demographics
The particular mutation responsible for pyruvate kinase deficiency differs between populations. For example, the 1529A mutation is believed to cause at least 1 gene mutation in most affected white persons from the United States and Europe. Other individuals, such as Portuguese, Spanish, and Italian persons, are affected by the 1456T, a mutation that impairs kinetic properties of the enzyme, resulting in a much milder clinical course . The authors have described a compound heterozygote for the known G1529A and a novel, previously undescribed frame shift mutation (1573 del T) whose clinical course was moderately severe, requiring several transfusions in the neonatal period.(ref43)(ref44). Asian persons in the United States appear to be affected by the 1468T mutation.
Age-related demographics
The age of onset for inherited pyruvate kinase deficiency correlates with severity. Persons with severe disease usually have onset in the neonatal period or infancy. In most affected persons, the disorder is detected during childhood, but in individuals who are mildly affected, it may not be detected until late adulthood.
Acquired pyruvate kinase deficiency is usually secondary to a particular disease. In such cases, the age of onset varies with the primary disease.
Prognosis
PKD is associated with a wide range of morbidities. Some patients present with a mild, compensated, chronic hemolytic anemia that does not require medical intervention; others present with a severe hemolytic anemia that, in most cases, requires only transfusions during childhood.
Morbidity and mortality correlate with disease severity and usually depend on complications. Mild and moderate forms of the disease are associated with an excellent prognosis. Severe forms of the disease are mostly symptomatic during early childhood. Following early childhood, the disorder is much better tolerated. Hydrops fetalis has been reported in a severely affected fetus. [28]
Morbidity and mortality
Complications associated with pyruvate kinase deficiency include the following:
-
Cholecystolithiasis is common in the first decade of life in children with severe anemia
-
Splenectomy increases the risk of sepsis by encapsulated bacteria in children and the risk of thromboembolic disease in adults [29]
-
Sudden worsening of anemia associated with viral infections (eg, parvovirus B19) can occur, leading to a transient decrease in red cell production (ie, aplastic crisis)
-
Severe anemia may result in heart failure
-
Ischemic stroke has been reported in previously undiagnosed young adults with pyruvate kinase deficiency [30]
-
Blood transfusions expose a person to the risk of contracting certain infections that are not well detected (eg, human immunodeficiency virus (HIV) disease, hepatitis C)
-
Repeated transfusions during pregnancy increase the risk of alloimmunization, which may lead to fetal complications
Morbidity in the newborn with pyruvate kinase deficiency is usually the result of severe anemia, hyperbilirubinemia, or both combined with the adverse effects associated with the management of such conditions. A report from the Netherlands described a fatal outcome in 2 newborns who suffered liver failure as a result of very severe pyruvate kinase–deficient hemolytic anemia. No other explanation for the liver failure was identified. [33]
Patient Education
Patients should understand their need for regular use of folic acid and vitamin B supplements and to avoid salicylates.
The risk regarding splenectomy versus that of multiple transfusions should be discussed with the parents of children with severe anemia. If a child has splenomegaly, parents should be instructed to have the child refrain from participating in contact sports.
Because the inheritance of pyruvate kinase deficiency is phenotypically autosomal recessive, parents and patients should be informed that the risk of recurrence is low.
-
Peripheral blood smear in a child with splenectomy and pyruvate kinase deficiency.
-
The Embden-Meyerhof pathway.
-
Pyruvate kinase in the Embden-Meyerhof pathway.







