Practice Essentials
Purine nueoside phosphorylase (PNP) deficiency causes a form of severe combined immunodeficiency (SCID) characterized by profound T cell deficiency, failure to thrive (FTT), recurrent deep seeded infection, developmental delay, progressive neurological deterioration, and autoimmune complications. Early diagnosis and early implementation of bone marrow transplantation (BMT) are crucial to minimize neurodevelopmental complications and ensure productive adult life in patients with PNP deficiency. However, one of the expected difficulties is that PNP deficiency can be missed with current newborn SCID screening measures. [1]
Background
Two genetic defects of the purine salvage pathway account for two immunodeficiencies that result in severe combined immunodeficiency (SCID). [2, 3] One disorder is adenosine deaminase (ADA) deficiency, which is Online Mendelian Inheritance in Man (OMIM) subject number 102700, and the other is purine nucleoside phosphorylase (PNP) deficiency, which is OMIM subject number 164050.
ADA and PNP deficiencies are autosomal recessive disorders. ADA and PNP are ubiquitous "housekeeping genes." In both disorders, the enzyme deficiencies result in accumulation of toxic metabolites, especially in lymphocytes. In ADA deficiency, the toxic metabolites block T-cell, B-cell, and natural killer (NK)-cell development; whereas in PNP deficiency, the metabolites are especially toxic to T-lineage cells, resulting in profound T-cell deficiency and variable degree of B-cell dysfunction.
In addition, both ADA and PNP deficiencies cause developmental delays and progressive neurological deterioration if not treated. This is especially prevalent in PNP deficiency with neurologic symptoms, including intellectual disability and muscle spasticity, reported in 67% of patients with PNP deficiency. In addition, PNP deficiency is associated with increased risk of autoimmune disorders, such as autoimmune hemolytic anemia, immune thrombocytopenia, neutropenia, thyroiditis, and lupus.
ADA deficiency results in absence of T, B, and NK cells, resulting in a SCID with marked lymphopenia. PNP deficiency causes profound T lymphopenia and variable numbers of B and NK cells. Serum immunoglobulin (Ig) levels are normal to near-normal, but specific antibody production is impaired.
Pathophysiology
PNP is an enzyme in the purine salvage pathway that metabolizes inosine and guanosine to hypoxanthine. [4, 5, 6, 7] In the preceding step of the pathway, ADA metabolizes adenosine to inosine. ADA deficiency causes a SCID that accounts for approximately 20% of all SCID cases. In both metabolic disorders, the enzyme deficiencies cause the accumulation of metabolites that are toxic to lymphoid lineage cells. See the image below.
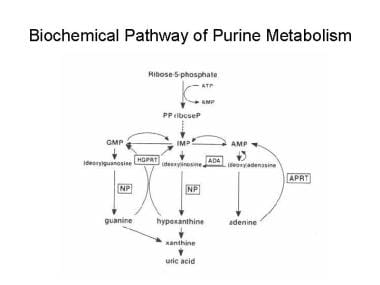 Biochemical pathway of purine metabolism. AMP = adenosine monophosphate, APRT = adenine phosphoribosyltransferase, GMP = guanosine monophosphate, HGPRT = hypoxanthine-guanine phosphoribosyltransferase, IMP = inosine monophosphate, NP = nucleoside phosphorylase, PPriboseP = 5-phosphorylribose-1-pyrophosphate.
Biochemical pathway of purine metabolism. AMP = adenosine monophosphate, APRT = adenine phosphoribosyltransferase, GMP = guanosine monophosphate, HGPRT = hypoxanthine-guanine phosphoribosyltransferase, IMP = inosine monophosphate, NP = nucleoside phosphorylase, PPriboseP = 5-phosphorylribose-1-pyrophosphate.
In adenosine deaminase deficiency, adenosine and adenine accumulate in the plasma. [8, 9] ATP accumulates in erythrocytes, and ADP, guanosine triphosphate (GTP), and ATP accumulate in lymphocytes. Deoxy-ATP (dATP) can reach toxic levels that inhibit ribonucleotide reductase, an enzyme essential for synthesis of DNA precursors.
In purine nucleoside phosphorylase deficiency, similar changes occur, resulting in elevated deoxy-GTP (dGTP) levels. dATP and dGTP predominantly accumulates in lymphoid tissue. dGTP inhibits ribonucleotide reductase, which is needed for synthesis of deoxynucleotides. In both adenosine deaminase and purine nucleoside phosphorylase deficiencies, thymocytes are thought to be selectively destroyed because of elevated levels of dATP and dGTP.
In a further description of the mechanism of T-cell depletion in purine nucleoside phosphorylase deficiency, Arpaia et al reported increased in vivo apoptosis of T cells and increased in vitro sensitivity to gamma irradiation in a murine model. [4] The immune deficiency in purine nucleoside phosphorylase deficiency may be the result of inhibited mitochondrial DNA repair due to the accumulation of dGTP in the mitochondria. The end result is increased sensitivity of T cells and thymocytes to spontaneous mitochondrial damage, leading to T-cell depletion due to apoptosis.
With adenosine deaminase deficiency, destruction of resting T cells and B cells is increased. In comparison, purine nucleoside phosphorylase deficiency results in selective destruction of T cells, with little effect on B cells. Numerous mutations of the ADA gene (on chromosome 20) and PNP genes (on band 14q13) have been identified. [2] Purine nucleoside phosphorylase is a trimer with molecular weight of 84-94 kDa. Most identified mutations are missense mutations, but deletion is also described. All reported patients with homozygous mutations of PNP have been symptomatic. Because only small amounts of adenosine deaminase are necessary for competent immunity, some patients with ADA mutations may still have 8-42% adenosine deaminase activity and no profound immunodeficiency. [2, 3]
Epidemiology
Frequency
Purine nucleoside phosphorylase deficiency is rare; only about 70 affected individuals have been described in the medical literature. This disorder accounts for approximately 4% of all SCID cases. [7, 10, 11]
Mortality/Morbidity
Patients with PNP deficiency are at risk for life-threatening recurrent viral, bacterial, fungal, mycobacterial, and protozoal infections. In addition, failure to thrive eventually ensues. The risk of lymphoma is also increased in patients with PNP deficiency. Neurologic symptoms, including intellectual disability and muscle spasticity, are major comorbid conditions that affect 67% of patients with PNP deficiency in one report and neurological deterioration is progressive with age if not treated.
Bone marrow transplantation may cure the immunodeficiency but does not reverse neurological damage that has already been caused by toxic metabolites. Patients are at risk for autoimmune diseases, including autoimmune hemolytic anemia, immune thrombocytopenia, thyroiditis, neutropenia, and lupus.
Demographics
PNP and ADA deficiencies are autosomal recessive disorders with equal incidence in boys and girls.
Although symptoms typically appear in the first year of life in patients with PNP deficiency, gradual deterioration of the T-cell immune system may delay the onset of symptoms until the second year of life. As above, neurological deterioration is also pregressive with age.
Prognosis
Bone marrow transplantation (BMT) can cure immunodeficiency and prevent neurological deterioration. Since neurodevelopmental impairment is progressive with age, early diagnosis and treatment (BMT) will be the key to minimize neurodevelopmental damage and other complications. However, PNP deficiency can be missed with current newborn SCID screening which assesses native T cell output from the thymus by measuring TCR rearrangement excision circle (TREC), since progressive loss of T cells in PNP deficiency due to toxic metabolites may not be fully manifested at birth. A high index of suspicion is necessary for early diagnosis of PNP to have the best clinical outcome.
The patient's prognosis depends on the success of immune reconstitution of the T-cell and B-cell systems. If immune reconstitution is successful, the patient's prognosis is good. However, bone marrow transplantation does not correct the neurologic disease.
Patient Education
Once diagnosis was made by measuring PNP activity, a patient and his/her family members need to be educated for treatment options (mainly BMT) and expected outcomes. Especially it needs to be clarified that immunodeficiency is curable by BMT but neurodevelopmental impairment caused by toxic metabolites may not be reversible by BMT.
-
Biochemical pathway of purine metabolism. AMP = adenosine monophosphate, APRT = adenine phosphoribosyltransferase, GMP = guanosine monophosphate, HGPRT = hypoxanthine-guanine phosphoribosyltransferase, IMP = inosine monophosphate, NP = nucleoside phosphorylase, PPriboseP = 5-phosphorylribose-1-pyrophosphate.
Tables
Study |
Infantile Onset |
Late Onset |
Adult Onset |
Lymphopenia |
Markedly decreased |
Decreased |
Decreased |
CD3+ cells |
Absent or trace |
Markedly reduced |
Markedly reduced |
CD4/CD8 ratio |
Too few to test |
< 1 |
< 1 |
Phytohemagglutinin response |
Absent |
Reduced |
Reduced |
Antigen response |
Absent |
Trace |
Trace |
Mixed lymphocyte culture response |
Reduced |
... |
... |
Ig response |
Absent |
Low to absent |
Normal (low IgG2) |
IgE |
Low |
Elevated |
Elevated |
Antibody response |
Absent |
Absent to low |
Low to polysaccharides antigens |
Eosinophilia |
Rare |
Common |
Common |
Infections |
Predominantly viral, fungal, opportunistic, bacterial |
Bacterial sinopulmonary |
Bacterial sinopulmonary, varicella-zoster, herpes simplex, candidal |
Brand (Manufacturer) |
Manufacturing Process |
pH |
Additives* |
Parenteral Form and Final Concentration |
IgA Content (mcg/mL) |
Carimune NF (CSL Behring) |
Kistler-Nitschmann fractionation; pH 4, nanofiltration |
6.4-6.8 |
6% solution: 10% sucrose < 20 mg NaCl/g protein |
Lyophilized powder 3%, 6%, 9%, 12% |
Trace |
Flebogamma (Grifols USA) |
Cohn-Oncley fractionation, polyethyline glycol (PEG) precipitation, ion-exchange chromatography, pasteurization |
5.1-6 |
Sucrose-free, contains 5% D-sorbitol |
Liquid 5% |
< 50 |
Gamunex (Talecris Biotherapeutics) |
Cohn-Oncley fractionation, caprylate-chromatography purification, cloth and depth filtration, low pH incubation |
4-4.5 |
Contains no sugar, contains glycine |
Liquid 10% |
46 |
Iveegam EN (Baxter Bioscience) |
Cohn-Oncley fraction II/III; ultrafiltration; pasteurization |
6.4-7.2 |
5% solution: 5% glucose, 0.3% NaCl |
Lyophilized powder 5% |
< 10 |
Gammagard S/D, Polygam S/D (Baxter Bioscience for the American Red Cross) |
Cohn-Oncley cold ethanol fractionation, cation and anion exchange chromatography, solvent detergent treated, nanofiltration, low pH incubation |
6.4-7.2 |
5% solution: 0.3% albumin, 2.25% glycine, 2% glucose |
Lyophylized powder 5%, 10% |
< 1.6 (5% solution) |
Gammagard Liquid 10% (Baxter Bioscience) |
Cohn-Oncley cold ethanol fractionation, cation and anion exchange chromatography, solvent detergent treated, nanofiltration, low pH incubation |
4.6-5.1 |
0.25M glycine |
Ready-for-use Liquid 10% |
37 |
Octagam (Octapharma USA) |
Cohn-Oncley fraction II/III; ultrafiltration; low pH incubation; S/D treatment pasteurization |
5.1-6 |
10% maltose |
Liquid 5% |
200 |
Panglobulin (Swiss Red Cross for the American Red Cross) |
Kistler-Nitschmann fractionation; pH 4, trace pepsin, nanofiltration |
6.6 |
Per gram of IgG: 1.67 g sucrose, < 20 mg NaCl |
Lyophilized powder 3%, 6%, 9%, 12% |
720 |
Privigen Liquid 10% (CSL Behring) |
Cold ethanol fractionation, octanoic acid fractionation, and anion exchange chromatography; pH 4 incubation and depth filtration |
4.6-5 |
L-proline (~250 mmol/L) as stabilizer; trace sodium; does not contain carbohydrate stabilizers |
Ready-for use liquid 10% |
< 25 |
*IVIG products containing sucrose are more often associated with renal dysfunction, acute renal failure, and osmotic nephrosis, particularly with preexisting risk factors (eg, history of renal insufficiency, diabetes mellitus, age >65 y, dehydration, sepsis, paraproteinemia, nephrotoxic drugs). |
|||||




