Practice Essentials
Porphyria is a predominantly inherited metabolic disorder resulting from a deficiency of an enzyme in the heme production pathway and overproduction of toxic heme precursors. Eight different enzymes are involved in the pathway, and deficiencies of the second to eighth enzyme result in a family of disorders with various, and often overlapping, clinical presentations.
Porphyrias are divided into two types according to the predominant symptoms: (1) the neurovisceral or acute porphyrias, with abdominal pain, neuropathy, autonomic instability, and psychosis, and (2) the cutaneous porphyrias, with symptoms of photosensitive lesions on the skin. [1, 2, 3, 4]
Aminolevulinic acid dehydrase (ALAD) porphyria and acute intermittent porphyria (AIP) cause predominately neurovisceral symptoms, whereas congenital erythropoietic porphyria (CEP), porphyria cutanea tarda (PCT), and erythropoietic porphyria (EP) cause mostly cutaneous symptoms. Two porphyrias overlap these categories and can cause both neurovisceral and cutaneous symptoms, namely hereditary coproporphyria (HCP) and variegate porphyria (VP). [5]
Only the cutaneous manifestations of the porphyrias are considered in this article. For explanation of diagnosis and management of the acute porphyrias and the acute manifestations of porphyrias with both neurovisceral and cutaneous components, please refer to the companion article Porphyria, Acute.
Some of the confusion with reference to the porphyrias is derived from the many synonyms for each particular disorder. [6] The synonyms are as follows:
Congenital erythropoietic porphyria (CEP)
See the list below:
-
Uroporphyrinogen III synthase deficiency
-
Hereditary erythropoietic porphyria
-
Congenital hematoporphyria
-
Erythropoietic uroporphyria
-
Gunther porphyria
Porphyria cutanea tarda (PCT)
See the list below:
-
Symptomatic porphyria
-
Uroporphyrinogen decarboxylase deficiency
Hepatoerythropoietic porphyria (HEP)
See the list below:
-
Homozygous type II PCT
Hereditary coproporphyria (HCP)
See the list below:
-
Coproporphyria
-
Coproporphyrinogen oxidase deficiency
Variegate porphyria (VP)
See the list below:
-
Protoporphyrinogen oxidase deficiency
-
South African porphyria
-
Porphyria variegata
-
Protocoproporphyria hereditaria
Erythropoietic protoporphyria (EPP)
See the list below:
-
Protoporphyria
-
Ferrochelatase deficiency
Signs of cutaneous porphyria
Skin changes are the hallmark of the cutaneous porphyrias. They can be acute (CEP), with erythema, edema, and erosions that eventually lead to facial scarring, or more chronic (PCT, VP, HCP), with skin fragility, blistering, and scarring, often over the backs of the hands. [7]
Workup in cutaneous porphyria
Demonstration of elevated porphyrins in plasma (particularly for congenital erythropoietic porphyria [CEP]), urine, and stool is very useful for diagnosis of the porphyrias. [8, 9]
Qualitative urine examination can identify urine porphyrins. However, normal urine contains porphyrins, making comparison with a control sample essential. Quantitative urine porphyrin levels can be useful, but prior qualitative testing is desirable.
Stool porphyrin levels that are combined with other laboratory values and clinic correlation help guide the diagnosis. However, levels of porphyrins widely vary, and, in most cases, exact values for each disorder have not been established.
Protoporphyria can be diagnosed by identifying numerous fluorescent erythrocytes in blood examined microscopically with a 100-watt iodine-tungsten lamp.
Management of cutaneous porphyria
Iron depletion can treat several of the cutaneous porphyrias. Phlebotomy and apheresis can remove excessive iron in patients with porphyria cutanea tarda (PCT). [10]
Porphyrin levels can be reduced by direct methods or with medications that bind porphyrins. These methods are useful adjuncts to iron load reduction therapy or when such therapy is ineffective or limited because of comorbid conditions, such as severe renal disease.
Oral photoprotection can be achieved with free radical scavengers, thereby reducing free radicals, singlet oxygen formation, and the photosensitizing effect of porphyrins.
Beta-carotene is a pigment found in various green and yellow fruits and vegetables and can decrease the severity of photosensitivity reactions in patients with porphyria. Sunscreen protection agents should be used if sun exposure is expected.
Cholecystectomy may be required for severe cholelithiasis in CEP. Splenectomy may be required if severe hemolytic anemia develops in CEP.
CEP has been cured with allogenic bone marrow transplant. Risks of this procedure must be carefully considered. Liver transplant alone is not curative for erythropoietic protoporphyria (EPP) but instead needs to be combined with bone marrow transplant. [11]
Pathophysiology
An outline of the porphyrin pathway reveals the pathophysiological mechanisms that cause porphyria. [12, 13, 14, 7]
Biosynthesis of one heme molecule requires 8 molecules of glycine and succinyl-coenzyme A (CoA). Heme is essential in many critical biochemical functions. For example, oxygen binding and transport, mixed-function oxidation in the cytochrome P-450 pathway, activation and decomposition of hydrogen peroxide, oxidation of tryptophan and prostaglandins, and the production of cyclic guanosine monophosphate (cGMP) cannot occur without heme.
The liver produces approximately 15% of the body's heme, but the majority is produced in the bone marrow. Heme produced in the liver is primarily used to produce cytochromes and peroxisomes, and heme produced in the bone marrow is primarily used for hemoglobin synthesis and oxygen transport.
As demonstrated in the following image, enzymes are located in either the mitochondria or the cytosol.
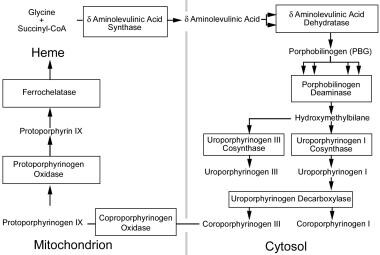 The heme production pathway. Heme production begins in the mitochondria, proceeds into the cytoplasm, and is then resumed in the mitochondria for the final steps. This figure outlines the enzymes and intermediates involved in the porphyrias. Enzymes names are presented in the boxes. Names of the intermediates are outside the boxes, between arrows. Multiple arrows leading to a box demonstrate that multiple intermediates are required as substrates for the enzyme to produce one product.
The heme production pathway. Heme production begins in the mitochondria, proceeds into the cytoplasm, and is then resumed in the mitochondria for the final steps. This figure outlines the enzymes and intermediates involved in the porphyrias. Enzymes names are presented in the boxes. Names of the intermediates are outside the boxes, between arrows. Multiple arrows leading to a box demonstrate that multiple intermediates are required as substrates for the enzyme to produce one product.
Delta-aminolevulinic acid (ALA) synthase is the first enzyme in the heme biosynthesis pathway. This enzyme condenses glycine and succinyl-CoA and has 2 isoforms that are encoded by separate genes; the housekeeping isoform is expressed in all tissues, whereas the erythroid isoform is expressed only in hematological tissue.
-
ALA synthase is the rate-limiting step for heme production in the liver but not the bone marrow. Indeed, the erythron responds to stimuli for heme synthesis by increasing cell numbers.
-
In the liver, ALA dehydratase and porphobilinogen (PBG) deaminase levels are typically low, resulting in ALA and PBG accumulation with increased ALA production under normal conditions.
-
High ALA levels induce heme oxygenase, increase bilirubin production, and inhibit ALA synthase.
-
Heme inhibits ALA synthase synthesis, mitochondrial transfer, and catalytic activity in the liver. This leads to tight control of ALA production because ALA synthase turnover is rapid.
-
Exogenous chemicals can induce ALA synthase by depleting existing heme or inhibiting heme synthesis. The 3 common mechanisms for this include the destruction or enhanced production of cytochrome P-450 heme and rapid inhibition of ferrochelatase.
-
In contrast to the liver, heme increases the synthesis of hemoglobin and ALA synthase in the bone marrow. In addition, erythroid ALA synthase is not affected by exogenous chemicals.
ALA dehydratase condenses 2 molecules of ALA to form the monopyrrole PBG ALA dehydratase, which is inhibited by lead, levulinic acid, hemin, succinylacetone, and alcohol.
-
Lead displaces zinc from the enzyme. This inhibition can be completely reversed by supplemental zinc or dithiothreitol.
-
Succinylacetone, a substrate analogue of ALA that is found in the blood and urine of patients with hereditary tyrosinemia, is the most potent inhibitor of ALA dehydratase.
PBG deaminase catalyzes the polymerization of 4 molecules of PBG in a head-to-tail orientation, yielding a linear tetrapyrrole intermediate, hydroxymethylbilane. The tissue and erythrocyte isozymes are encoded by the same structural gene.
Uroporphyrinogen III cosynthase forms uroporphyrinogen III from hydroxymethylbilane by reversing the orientation of the last pyrrole ring before cyclizing the linear molecule. Uroporphyrinogen I cosynthase forms uroporphyrinogen I from hydroxymethylbilane by cyclizing the linear molecule without modifying any of the pyrrole rings. Normal tissues contain an excess of uroporphyrinogen cosynthases compared to PBG deaminase.
Uroporphyrinogen decarboxylase sequentially removes a carboxylic group from the acetic side chains of each of the pyrrole rings to yield coproporphyrinogen. This enzyme has highest affinity for uroporphyrinogen III. It is inhibited by several metals, including copper, mercury, and platinum, but the evidence indicating that iron has an effect on this enzyme is controversial.
Coproporphyrinogen oxidase removes a carboxyl group from the propionic groups on 2 of the pyrrole rings to yield protoporphyrinogen IX.
Protoporphyrinogen oxidase forms protoporphyrin by removing 6 hydrogen atoms from protoporphyrinogen IX. This enzyme has been identified in human fibroblasts, erythrocytes, and leukocytes. It is noncompetitively and irreversibly inhibited by hemin.
Iron is inserted into protoporphyrin by ferrochelatase as the last step in the heme synthesis pathway. Enzyme activity is stimulated by fatty acids and is inhibited by metals, such as cobalt, zinc, lead, copper, and manganese, as well as by metalloporphyrins.
Porphyria cutanea tarda
Porphyrin overproduction occurs in the liver and the skin. Singlet oxygen, which is the primary toxic agent in the photodermatoses of porphyria, is the high-energy form of oxygen in which all the outer shell electrons are paired. [15] It is generated by visible light (400 nm) in the presence of photosensitizers, such as the various porphyrins. [16] Abnormally high complement and prostaglandins occur in lesions.
Erythropoietic protoporphyria
Mechanisms are similar to PCT, except that locally cutaneous production of porphyrins probably does not occur. [17]
Congenital erythropoietic porphyria
Bone marrow is the primary site of the enzyme defect. [18, 19] Conspicuous porphyrin-laden normoblasts and reticulocytes are found in the marrow. Photolysis of porphyrin-laden erythrocytes occurs in the dermal capillaries, causing subepidermal lesions. Repeated trauma causes secondary skin changes and results in joint contractures. An intrinsic erythrocyte abnormality results in autohemolysis.
Splenomegaly occurs as a consequence of the removal of damaged and hemolyzed erythrocytes. Cholecystitis results from porphyrin-rich gallstones. Bone marrow hyperexpansion and vitamin D deficiency due to avoiding sun exposure result in fragile bones.
Epidemiology
Frequency
United States
The absence of a porphyria registry in the United States impedes an accurate calculation of frequencies, but the overall prevalence is estimated as 4 per 100,000. However, as indicated in Table 2, the porphyria incidence varies significantly by type, with PCT being the most common and CEP being very rare. [20] The lack of recognition of these disorders may contribute to inaccurate knowledge of their true incidence.
Despite earlier reports, the frequency of the genetic defect and phenotypic expression have a moderately strong relationship. A highly variable penetrance rate has been noted. Expression of the genetic defect is more common in familial cases, suggesting that such families may have an additional undetected genetic abnormality or environmental exposure. Around one half of individuals with genetic defects are symptomatic.
International
In general, porphyrias do not have a geographic preference. However, certain porphyrias have a high incidence in certain parts of the world. [21]
PCT type I (ie, sporadic) is more common than PCT types II and III (ie, familial) in Europe, South Africa, and South America. Incidence of HCP is widely varies by race. [22] Incidence of VP is particularly high in South Africans of Danish descent. [23]
Table 1. Frequency Varies with the Specific Porphyria (Open Table in a new window)
Type of Porphyria |
Age of Onset |
Incidence per 100,000 Population |
Male-to-Female Ratio |
CEP |
Infancy to early childhood; rare in adults |
300 cases total |
1:1 |
PCT |
Type I: Adulthood Type II (heterozygous mutations): Adulthood Type III (homozygous mutations): Childhood |
United States: 4 United Kingdom: 0.05 |
1:1 |
HCP |
Predominantly adulthood Youngest report was child aged 4 y |
Japan: 1.5 Czech: 1.5 Israel: 0.7 Denmark: 0.05 |
1:20 1:4 2:1 1:1 |
VP |
Heterozygous mutation: After puberty Homozygous mutation: Childhood (rare) |
South Africa: 34 |
1:1 |
EPP |
Infancy to childhood |
0.02 |
1:1 |
Mortality/Morbidity
CEP is associated with a significant decrease in life expectancy.
Race
PCT does not have a racial predilection, except in South Africa, where it is more prevalent among persons of Bantu origin. This is believed to be caused by a higher incidence of hemosiderosis in these individuals.
The incidence of HCP greatly depends on race.
VP has a particularly high incidence in South African persons of Dutch descent.
See International.
Sex
Most porphyrias do not demonstrate a sex predilection (see International).
A change to a nearly equal sex distribution is attributed to the higher rate of alcoholism in males combined with the recent increase in the use of estrogens by women. Both factors exacerbate manifestations of the porphyrias.
The sex predilection of HCP varies with race (see International).
Age
CEP and EP usually present in infancy, but manifestations can be delayed until childhood. CEP can cause hydrops fetalis and recurrent fetal loss.
HCP has a variable age of onset but usually does not present before adolescence. However, cases have been reported at younger ages.
Symptoms of PCT and VP most often manifest in adulthood. However, inheritance of 2 abnormal genes can cause onset in childhood. Childhood onset is more unusual for VP than PCT, and, in the case of PCT, onset in infancy has been reported. Because PCT has a relatively high prevalence and low penetrance, 2 asymptomatic carriers each can transmit an abnormal gene without knowledge of the existing abnormality. HEP represents the onset of homozygous PCT type II in childhood.
-
The heme production pathway. Heme production begins in the mitochondria, proceeds into the cytoplasm, and is then resumed in the mitochondria for the final steps. This figure outlines the enzymes and intermediates involved in the porphyrias. Enzymes names are presented in the boxes. Names of the intermediates are outside the boxes, between arrows. Multiple arrows leading to a box demonstrate that multiple intermediates are required as substrates for the enzyme to produce one product.
Tables
Type of Porphyria |
Age of Onset |
Incidence per 100,000 Population |
Male-to-Female Ratio |
CEP |
Infancy to early childhood; rare in adults |
300 cases total |
1:1 |
PCT |
Type I: Adulthood Type II (heterozygous mutations): Adulthood Type III (homozygous mutations): Childhood |
United States: 4 United Kingdom: 0.05 |
1:1 |
HCP |
Predominantly adulthood Youngest report was child aged 4 y |
Japan: 1.5 Czech: 1.5 Israel: 0.7 Denmark: 0.05 |
1:20 1:4 2:1 1:1 |
VP |
Heterozygous mutation: After puberty Homozygous mutation: Childhood (rare) |
South Africa: 34 |
1:1 |
EPP |
Infancy to childhood |
0.02 |
1:1 |
Porphyria |
Deficient Enzyme |
Location |
Inheritance |
Chromosome Band |
CEP |
Uroporphyrinogen III synthase |
Cytosol |
Autosomal recessive (AR) |
10q25.3-26.3 |
PCT |
Uroporphyrinogen decarboxylase |
Cytosol |
Autosomal dominant (AD) |
1p34 |
HEP |
Uroporphyrinogen decarboxylase |
Cytosol |
AR |
1p34 |
HCP |
Coproporphyrinogen oxidase |
Mitochondrial |
AD |
3q12 |
VP |
Protoporphyrinogen oxidase |
Mitochondrial |
AD |
1q22-23 |
EPP |
Ferrochelatase |
Mitochondrial |
AD, AR |
18q22 |
Porphyrin Type |
CEP |
PCT |
HCP |
VP |
EPP |
Uroporphyrin |
Significantly increased |
Increased |
Within reference range |
Within reference range |
Within reference range |
Coproporphyrin |
Significantly increased |
Increased |
Significantly increased |
Increased |
Within reference range |
Protoporphyrin |
Within reference range |
Within reference range |
Increased |
Significantly increased |
Significantly increased |
Porphyrin type |
CEP and PCT |
HCP and VP |
5-Aminolevulinate |
Within reference range |
Significantly increased |
PBG |
Within reference range |
Significantly increased |
Uroporphyrin |
Significantly increased |
Increased |
Coproporphyrin |
Increased |
Significantly increased |







