Background
Glycogen-storage disease type II (GSD II), also known as Pompe disease, is part of a group of metabolic diseases called lysosomal storage disorders (LSDs). [1] GSD II is an autosomal-recessive disorder that results from deficiency of acid alpha-glucosidase (also known as acid maltase), a lysosomal hydrolase. The cellular role of acid alpha-glucosidase is to convert glycogen into glucose within the lysosomes. The Danish pathologist Joannes Cassianus Pompe first described this disease in 1932 when he was presented with a 7-month-old girl who died after developing idiopathic hypertrophic cardiomyopathy. Pompe observed an abnormal accumulation of glycogen in all postmortem tissues examined and described the cardinal pathologic features of this lysosomal storage disorder.
Pompe disease (GSD II) has a broad clinical spectrum. Classification is based on age of onset, organ involvement, severity, and the rate of disease progression. Three major forms of GSD II are recognized: classic infantile-onset, non-classic variant of infantile-onset, and late-onset (includes childhood, juvenile, and adult-onset).
Classic infantile-onset Pompe disease may be apparent in utero, but more often presents within the first 2 months of life. In this form, cardiac, skeletal, and respiratory muscles are involved. Clinical hallmarks of classic infantile-onset Pompe disease include hypotonia, generalized muscle weakness, cardiomegaly, hypertrophic cardiomyopathy, feeding difficulties, failure to thrive, respiratory distress, and hearing loss. Classic infantile-onset GSD II is marked by a progressive and rapidly fatal course. Without enzyme replacement therapy (ERT), classic infantile-onset Pompe disease commonly results in death within the first year of life due to cardiac disease from progressive left ventricular (LV) outflow obstruction.
The non-classic variant of infantile-onset Pompe disease presents within the first year of life. Patients older than 6 months present with motor delays and/or slowly progressive muscle weakness. Death results from ventilatory failure in early childhood. In this form, cardiac disease is not found to be a major cause of morbidity; however, cardiomegaly may be seen.
Late-onset GSD II is characterized by proximal muscle weakness and respiratory compromise. Adults with late-onset GSD II typically present with proximal muscle weakness between the second and sixth decades of life. These individuals ultimately die of respiratory failure. Cardiac involvement is less likely among individuals with disease onset at an older age. [2, 3]
The photomicrographs below document liver findings in GSD II.
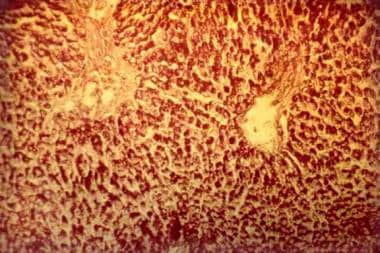 Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).
Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).
 Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).
Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).
Pathophysiology
Acid alpha-glucosidase (also known as acid maltase) is a lysosomal hydrolase that is required for the degradation of a small percentage (1-3%) of cellular glycogen. The main pathway for glycogen degradation is not deficient in GSD II; therefore, energy production is not impaired, and hypoglycemia does not occur. However, the deficiency of acid alpha-glucosidase activity does result in the accumulation of structurally normal glycogen in lysosomes and in the cytoplasm of affected individuals. Excessive glycogen storage within lysosomes interrupts normal functioning of other organelles and leads to cellular injury. In turn, this leads to enlargement and dysfunction of the entire organ involved (eg, cardiomyopathy).
In the classic infantile form of Pompe disease, clinically significant glycogen storage occurs in cardiac muscle. Over time, cardiomegaly with LV thickening occurs, eventually leading to outflow tract obstruction. Glycogen storage in skeletal muscle leads to hypotonia and weakness. The respiratory muscles are also affected, resulting in hypoventilation and progressive respiratory compromise. Central nervous system (CNS) involvement is primarily limited to the anterior horn cells of the spinal cord and brain stem nuclei. Despite CNS involvement, intellect remains normal. In non-classic variant infantile-onset Pompe disease, skeletal and respiratory involvement is common; the level of cardiac involvement varies. In cases of late-onset GSD II, significant cardiac involvement is not observed.
Epidemiology
The combined incidence of all forms of Pompe disease (GSD II) varies depending on ethnicity and geographic region.
United States
The total incidence of Pompe disease (all variants of GSD II) is estimated at 1 per 40,000 population. This calculation is based on estimated gene frequencies in healthy individuals from various US ethnic groups. The highest incidence has been observed in the African-American population, in which the combined incidence may be as high as 1 per 14,000 population. [4]
International
Among individuals of European descent, the incidence of infantile-onset Pompe disease is reported as 1 per 100,000 population and the incidence of late-onset Pompe disease is reported as 1 per 60,000 population. [5]
The combined incidence of Pompe disease in Taiwan and southern China is estimated at 1 per 50,000 population. [6]
The combined incidence of Pompe disease in the Netherlands is estimated at 1 per 40,000 population (1 per 138,000 population for infantile-onset, 1 per 57,000 population for late-onset [adult]). [7] A common GAA pathogenic variant among the Dutch people is c.525delT, which is found in 34% of cases. [8]
In Portugal, the combined incidence of Pompe disease is estimated at 1 per 600,000 population. [9]
In Australia, the combined incidence of Pompe disease is estimated at 1 per 145,000 population. [10]
Mortality/Morbidity
Classic infantile-onset GSD II usually is fatal during the first year of life. As weakness progresses, affected individuals develop feeding difficulties and respiratory insufficiency. Cardiac enlargement of the LV leads to outflow tract obstruction and ventricular failure. Death results from cardiopulmonary failure. Non-classic infantile-onset Pompe disease presents later, within the first year of life, with clinical findings of motor delays and progressive muscle weakness. Death due to ventilatory failure occurs in early childhood.
Late-onset Pompe disease (childhood and juvenile) progresses more slowly and is uniformly fatal. Affected individuals generally do not survive beyond the second or third decade of life. All affected individuals have involvement of pulmonary system, and most die of respiratory failure. Several patients with this form are reported to have died of basilar artery aneurysm [11] ; all were found to have abnormal glycogen storage within the lysosomes of arterial smooth muscle fibers.
Patients with late-onset (adult) Pompe disease may survive for decades following diagnosis. Muscle weakness may interfere with normal daily activities, and respiratory insufficiency is often associated with sleep apnea. Death usually results from respiratory failure.
Race
Common mutations within the GAA gene associated with the infantile-onset forms of Pompe disease have been discovered in Taiwanese, Dutch, and African-American populations. A common mutation associated with the late-onset form has been found in Whites.
Sex
GSD II has no sexual predilection.
GSD II is inherited as an autosomal recessive disorder with the gene locus at 17q25.3; hence, the inheritance of this disease is not related to the sex chromosomes.
Age
Age of onset helps distinguish the current three defined age-onset categories of GSD II. The classic and non-classic infantile-onset forms present from birth to 2 months of life and within the first year of life, respectively. The late-onset definition includes childhood, juvenile, and adult.
-
Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the intensively stained vacuoles in the hepatocytes (periodic acid-Schiff, original magnification X 27).
-
Glycogen-storage disease type II (Pompe disease). Photomicrograph of the liver. Note the regular reticular net and hepatocytes vacuolization (Gordon-Sweet stain, original magnification X 25).






