Practice Essentials
Polycythemia vera (PV) is a disorder of the multipotent hematopoietic stem cell that manifests as excess production of normal erythrocytes and variable overproduction of leukocytes and platelets. It is grouped with the Philadelphia chromosome–negative myeloproliferative disorders and can usually be differentiated from them by the predominance of erythrocyte production. [1] (See the image below.) Once polycythemia vera is suspected, the first step in evaluating a patient is determining whether the patient has primary, secondary, or apparent polycythemia. Treatment of the disease depends on whether it is in the plethoric phase or the spent phase.
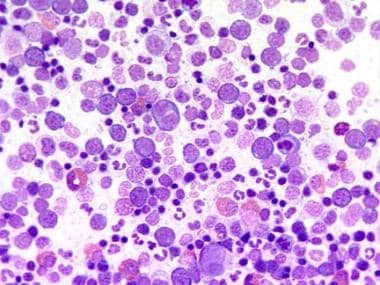 Bone marrow film at 400X magnification demonstrating dominance of erythropoiesis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Bone marrow film at 400X magnification demonstrating dominance of erythropoiesis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.
Signs and symptoms of polycythemia vera
Some patients with polycythemia vera are asymptomatic, whereas others have various nonspecific symptoms. Thirty percent of patients report headache, weakness, dizziness, and sweating (in order of decreasing frequency). Many of these symptoms can be attributed to excess hematocrit.
Alternatively, the patient may present with a complication of polycythemia vera. Approximately 33% of patients present with thrombosis or hemorrhage; 75% of these have arterial thrombosis, and 25% have venous thrombosis.
Potential physical findings include plethora and ruddiness of the face, erythromelalgia of the distal extremities, bruising, and splenomegaly. Specific attention should be directed towards sternal tenderness, which may indicate transformation to acute myeloid leukemia. [2]
Workup in polycythemia vera
Laboratory studies
Once polycythemia vera is suspected, the first step in evaluating a patient is determining whether the patient has primary, secondary, or apparent polycythemia.
A complete blood count (CBC), arterial blood gas (ABG) measurement, venous blood gas (VBG), and erythropoietin level can be used to differentiate patients.
Ferritin levels may also help to differentiate between primary and secondary polycythemias. Typically in primary polycythemia, the ferritin level is low due to constant overproduction of erythrocytes. In contrast, the ferritin level is usually normal in secondary polycythemia.
Imaging studies
Computed tomography (CT) scanning or ultrasonography of the abdomen can be used to assess the size of the spleen, which is frequently enlarged in polycythemia vera.
Histologic findings
Bone marrow and aspirate in polycythemia vera tend to be hypercellular. Some evidence of myelofibrosis may also be present.
In the plethoric phase, the blood smear shows normal erythrocytes, variable neutrophilia with myelocytes, metamyelocytes, and varying degrees of immaturity, basophilia, and increased platelets.
In the spent phase, the blood smear shows abundant teardrop cells, leukocytosis (or leukopenia), and thrombocytosis (or thrombocytopenia).
Management of polycythemia vera
Treatment of polycythemia vera depends on whether the disease is in the plethoric phase or the spent phase.
In the plethoric phase, the goal of treatment is controlling thrombotic episodes by restraining monoclonal proliferation rather than restoring polyclonal growth and maturation of cells. Interferon alfa is an exception; a few case reports have reported restoration of polyclonality.
In the plethoric phase, polycythemia vera is treated first by performing phlebotomy until the hematocrit is under reasonable control. Most patients can tolerate removal of 450-500 mL of blood every 2-4 days. As more blood is removed and the patient becomes iron deficient, the hematocrit becomes easier to control, and the phlebotomy schedule should be adjusted accordingly.
Risk stratification is important in deciding whether or not chemotherapeutic cytoreductive therapy is indicated.
Because of the theoretically possible leukemogenic risk of hydroxyurea, anagrelide has been used to control increased platelet counts, with the aim of reducing thrombotic events. In the PT1 trial in the United Kingdom, patients with essential thrombocythemia were randomly assigned to receive hydroxyurea or anagrelide. The study demonstrated an increased risk of thrombosis with anagrelide. The implication for polycythemia vera is unclear, but a reduction in platelet count does not affect rate of thrombosis in essential thrombocytopenia; a similar result is expected in polycythemia vera.
Pathophysiology
Clonality and erythropoietin (Epo) independence are the two key aspects of polycythemia biology. A single clonal population of erythrocytes, granulocytes, platelets, and variable clonal B cells arises when a hematopoietic stem cell gains a proliferative advantage over other stem cells. The T lymphocytes and natural killer cells remain polyclonal in polycythemia vera; this is related to their longevity. Clonality can currently only be tested in females using X-chromosome polymorphisms that take advantage of X-chromosome inactivation.
Epo independence is the ability of polycythemia vera hematopoietic stem cells to grow erythroid colonies without Epo. Although the colonies do not require erythropoietin, they remain responsive to it, and the erythropoietin receptor (EpoR) is normal, without defects in its function or quantity. Experiments using antibodies to neutralize Epo or block the EpoR do not abolish Epo-independent erythroid colony formation.
The understanding of the molecular mechanisms underlying polycythemia vera has been greatly enhanced over last decade. Genome-wide scanning that compared clonal polycythemia vera and nonclonal cells from the same individuals revealed a loss of heterozygosity (LOH) in chromosome 9p. This is found in approximately 30% of patients with polycythemia vera. This is not a classical chromosomal deletion but rather a duplication of a portion of a chromosome and the loss of the corresponding parental region. This process is called uniparental disomy. [3]
The 9p region contains a gene that encodes for the JAK2 tyrosine kinase. The JAK family of kinases is critical in cytokine receptor signaling and transmits the activating signal in the Epo-EpoR signaling pathway. Inhibition of JAK2 has been shown to eliminate Epo independence of erythroid progenitors. Subsequently, these observations were followed by the identification of a loss-of-function somatic mutation in an auto-inhibitory JAK2 domain, which essentially produces a gain-of-function mutation that affects the kinase. This occurs when a point mutation in exon 14 leads to a valine-to-phenylalanine mutation at codon 617 of the JAK2 gene. [4, 5]
The JAK2V617F mutation leads to constitutive phosphorylation, thus constitutive activity and STAT recruitment, which provides the proliferative advantage seen in polycythemia vera. This process occurs in the absence of Epo and explains both the Epo independence and Epo hypersensitivity of polycythemia vera colonies. A mouse model of this mutation produced a clinical phenotype consistent with polycythemia vera. These data were rapidly confirmed by several groups; each reported that more than 90% of patients with polycythemia vera carry the JAK2V617F mutation. [4] However, compelling data strongly argue that this mutation is not a disease-initiating mutation. [6] Rather, an as-of-yet unidentified mutation or mutations predispose to the acquisition of polycythemia vera. Patients with the JAK2V617F mutation tend to have the clinical phenotype of essential thrombocythemia.The quantitative allele burden (ratio of mutant to wild type expression) of JAK2V617F also has a clinical impact. Data from Vannucchi et al reveal that higher quantitative levels of the JAK2V617F allele correlated with higher values for hematocrit. [7] WBC and lactate dehydrogenase levels were positively correlated with the level of the mutation. The highest JAK2V617F levels at diagnoses predicted patients more likely to have splenomegaly, develop presenting pruritus, or eventually require chemotherapy.
Also, the rate of presenting major thromboses was positively correlated with higher mutation values. In fact, a multivariate analysis that included age, leukocytosis, hematocrit, platelet count, and therapies indicated that JAK2V617F/JAK2 wild type ratio is an independent risk factor for major vascular events. This was also validated by Silver et al, suggesting greater JAK2V617F allele burden correlates with higher white cell count, splenomegaly, and thromboembolic disease. [8, 9] They also suggested a higher frequency of myelofibrosis.
Carobbio et al demonstrated that the JAK2V617F allelic burden in JAK2V617F-positive essential thrombocythemia and polycythemia vera is the only risk factor that correlated with increased vascular events 5 years after diagnosis. [10]
A study by Szuber et al indicated that patients aged 40 years or younger with polycythemia vera are more likely than older patients with the disease to demonstrate a normal karyotype. [11]
Epidemiology
Frequency
United States
The incidence of polycythemia vera is reported to be 4.9 cases per 100,000 population in Baltimore. A more recent review of polycythemia vera in Connecticut reported an incidence of 22 cases per 100,000 population.
International
The incidence of polycythemia vera is reported to be 6.7 cases per 1,000,000 population in Israel, and reviews have estimated 30 cases per 100,000 population in Sweden and Italy. In Norway, the prevalence of polycythemia vera is reported to be 9.2 cases per 1,000,000 inhabitants. [12]
Race
The disease appears more common in Jews of European extraction than in most non-Jewish populations. Some familial forms of polycythemia vera are noted, but the mode of inheritance is not clear.
Sex
Men are preferentially affected over women. The male-to-female ratio is 1.2-2.2:1.
Age
Onset is typically in the sixth decade, and the peak incidence is at age 60-80 years. In the previously mentioned study by Szuber et al, the incidence of polycythemia vera in patients aged 40 years or younger with myeloproliferative neoplasms was 12%. [11]
Prognosis
The course of polycythemia vera may or may not follow two phases. The plethoric phase usually occurs first and is characterized by hyperproliferation of cellular components. The principle manifestations during this phase are thrombosis and hemorrhage. Consequently, treatment is aimed at ameliorating symptoms. The plethoric phase can last for a few years to as many as 20. Following the plethoric phase, the spent phase is characterized by progressive anemia, fibrosis, and splenomegaly. The smear demonstrates anemia, thrombocytosis (or thrombocytopenia), and leukocytosis (or leukopenia/neutropenia). In contrast to the plethoric phase, patients in the spent phase are often transfusion dependent.
The aforementioned study by Szuber et al found the incidence of fibrotic progression in polycythemia vera to be higher in patients aged 40 years or younger (22%) than in older patients, a phenomenon the investigators attributed to the longer survival period in the younger group. [11]
Patients are at risk for leukemic transformation throughout the entire course of disease although the rate is higher during the spent phase. The incidence of leukemia was found by the Polycythemia Vera Study Group (PVSG) to be affected by the mode of treatment. [13] Treatment with phlebotomy only, Phosphorus-32 (P32), and chlorambucil resulted in a leukemic incidence of 1.5%, 10%, and 13% respectively. The study by Szuber et al determined that over a median follow-up period of 11.3 years, 4% of patients aged 40 years or younger with polycythemia vera underwent documented leukemic transformation. [11]
Except for potential leukemic transformation, appropriately treated polycythemia vera is compatible with near normal life. Without treatment, 50% of patients die within 18 months of diagnosis, usually from a thrombotic event. Survival with treatment depends on modality. Median survival is 13.9 years for phlebotomy alone, 11.8 years for P32, and 8.9 years for chlorambucil.
In the study by Szuber et al, the investigators found the median period of survival for polycythemia vera patients aged 40 years or younger to be 37 years, compared with 22 years for patients aged 41-60 years, and 10 years for patients over age 60 years. [11]
A European study, by Marchioli and colleagues, attempted to further define the prognosis of this disease; [14] 1,638 patients were prospectively followed in an attempt to describe the clinical history of polycythemia vera. The primary limitation of this study is a mean follow-up of 2.7 years. The overall mortality rate was 3.7 death per 100 persons per year. This was primarily caused by a moderate rate of cardiovascular death (1.7 deaths per 100 persons per year) and a high rate of death from noncardiovascular causes (1.8 deaths per 100 persons per year), primarily hematologic transformations. Cardiovascular mortality accounted for 45% of all deaths. Hematologic transformation (13% of all deaths) and solid tumors (19.5%) were also significant causes of mortality.
As previously seen in other studies, age older than 65 years and history of previous thrombosis were also significantly associated with mortality risk. Cumulative rate of cardiovascular events was 5.5 per 100 persons per year. Rates of combined malignancy, hematologic transformation, and non–polycythemia vera related malignancies were 3, 1.3, and 1.7 per 100 persons per year, respectively.
-
Bone marrow film at 400X magnification demonstrating dominance of erythropoiesis. Courtesy of U. Woermann, MD, Division of Instructional Media, Institute for Medical Education, University of Bern, Switzerland.