Kim SY. Diagnosis and Treatment of Hypopituitarism. Endocrinol Metab (Seoul). 2015 Dec. 30 (4):443-55. [QxMD MEDLINE Link]. [Full Text].
Gounden V, Jialal I. Hypopituitarism (Panhypopituitarism). 2018 Jan. [QxMD MEDLINE Link]. [Full Text].
Alexandraki KI, Grossman A. Management of Hypopituitarism. J Clin Med. 2019 Dec 5. 8 (12):[QxMD MEDLINE Link]. [Full Text].
Migliaretti G, Aimaretti G, Borraccino A, et al. Incidence and prevalence rate estimation of GH treatment exposure in Piedmont pediatric population in the years 2002-2004: Data from the GH Registry. J Endocrinol Invest. 2006 May. 29(5):438-42. [QxMD MEDLINE Link].
Hanna CE, Krainz PL, Skeels MR, Miyahira RS, Sesser DE, LaFranchi SH. Detection of congenital hypopituitary hypothyroidism: ten-year experience in the Northwest Regional Screening Program. J Pediatr. 1986 Dec. 109(6):959-64. [QxMD MEDLINE Link].
Kao KT, Stargatt R, Zacharin M. Adult Quality of Life and Psychosocial Outcomes of Childhood Onset Hypopituitarism. Horm Res Paediatr. 2015. 84 (2):94-101. [QxMD MEDLINE Link].
Matthai SM, Smith CS. Pituitary hypoplasia associated with a single central maxillary incisor. J Pediatr Endocrinol Metab. 1996 Sep-Oct. 9(5):543-4. [QxMD MEDLINE Link].
Willnow S, Kiess W, Butenandt O, et al. Endocrine disorders in septo-optic dysplasia (De Morsier syndrome)--evaluation and follow up of 18 patients. Eur J Pediatr. 1996 Mar. 155(3):179-84. [QxMD MEDLINE Link].
Burgner DP, Kinmond S, Wallace AM, et al. Male pseudohermaphroditism secondary to panhypopituitarism. Arch Dis Child. 1996 Aug. 75(2):153-5. [QxMD MEDLINE Link].
Setian N, Aquiar CH, Galvao JA. Rathke's cleft cyst as a cause of growth hormone deficiency and micropenis. Child's Nervous System. 1999. Vol 5: 271-3.
Rajaratnam S, Seshadri MS, Chandy MJ, Rajshekhar V. Hydrocortisone dose and postoperative diabetes insipidus in patients undergoing transsphenoidal pituitary surgery: a prospective randomized controlled study. Br J Neurosurg. 2003 Oct. 17(5):437-42. [QxMD MEDLINE Link].
Borchert M, Garcia-Filion P. The syndrome of optic nerve hypoplasia. Curr Neurol Neurosci Rep. 2008 Sep. 8(5):395-403. [QxMD MEDLINE Link].
Rosenbloom AL, Almonte AS, Brown MR, et al. Clinical and biochemical phenotype of familial anterior hypopituitarism from mutation of the PROP1 gene. J Clin Endocrinol Metab. 1999 Jan. 84(1):50-7. [QxMD MEDLINE Link].
Ward L, Chavez M, Huot C, et al. Severe congenital hypopituitarism with low prolactin levels and age- dependent anterior pituitary hypoplasia: a clue to a PIT-1 mutation. J Pediatr. 1998 Jun. 132(6):1036-8. [QxMD MEDLINE Link].
Vieira TC, Boldarine VT, Abucham J. Molecular analysis of PROP1, PIT1, HESX1, LHX3, and LHX4 shows high frequency of PROP1 mutations in patients with familial forms of combined pituitary hormone deficiency. Arq Bras Endocrinol Metabol. 2007 Oct. 51(7):1097-103. [QxMD MEDLINE Link].
Castinetti F, Reynaud R, Saveanu A, et al. MECHANISMS IN ENDOCRINOLOGY: An update in the genetic aetiologies of combined pituitary hormone deficiency. Eur J Endocrinol. 2016 Jun. 174 (6):R239-47. [QxMD MEDLINE Link]. [Full Text].
Parkin K, Kapoor R, Bhat R, Greenough A. Genetic causes of hypopituitarism. Arch Med Sci. 2020. 16 (1):27-33. [QxMD MEDLINE Link]. [Full Text].
van Aken MO, Lamberts SW. Diagnosis and treatment of hypopituitarism: an update. Pituitary. 2005. 8(3-4):183-91. [QxMD MEDLINE Link].
Wijnen M, van den Heuvel-Eibrink MM, Janssen JAMJL, et al. Very long-term sequelae of craniopharyngioma. Eur J Endocrinol. 2017 Jun. 176 (6):755-67. [QxMD MEDLINE Link].
Bettendorf M, Fehn M, Grulich-Henn J, et al. Lymphocytic hypophysitis with central diabetes insipidus and consequent panhypopituitarism preceding a multifocal, intracranial germinoma in a prepubertal girl. Eur J Pediatr. 1999 Apr. 158(4):288-92. [QxMD MEDLINE Link].
Maghnie M, Genovese E, Sommaruga MG, et al. Evolution of childhood central diabetes insipidus into panhypopituitarism with a large hypothalamic mass: is 'lymphocytic infundibuloneurohypophysitis' in children a different entity?. Eur J Endocrinol. 1998 Dec. 139(6):635-40. [QxMD MEDLINE Link].
Mikami-Terao Y, Akiyama M, Yanagisawa T, et al. Lymphocytic hypophysitis with central diabetes insipidus and subsequent hypopituitarism masking a suprasellar germinoma in a 13-year-old girl. Childs Nerv Syst. 2006 Mar 25. [QxMD MEDLINE Link].
Tanriverdi F, Senyurek H, Unluhizarci K, et al. High risk of hypopituitarism after traumatic brain injury: a prospective investigation of anterior pituitary function in the acute phase and at 12-months after the trauma. J Clin Endocrinol Metab. 2006 Mar 7. [QxMD MEDLINE Link].
Behan LA, Phillips J, Thompson CJ, Agha A. Neuroendocrine disorders after traumatic brain injury. J Neurol Neurosurg Psychiatry. 2008 Jul. 79(7):753-9. [QxMD MEDLINE Link].
Auble BA, Bollepalli S, Makoroff K, et al. Hypopituitarism in pediatric survivors of inflicted traumatic brain injury. J Neurotrauma. 2014 Feb 15. 31 (4):321-6. [QxMD MEDLINE Link]. [Full Text].
You W, Zhu Y, Wen L, Sun Y, Pan D, Yang X. Risk Factors for Anterior Hypopituitarism in Patients With Traumatic Brain Injury. J Craniofac Surg. 2019 Oct. 30 (7):2119-23. [QxMD MEDLINE Link].
Lin Y, Hansen D, Sayama CM, Pan IW, Lam S. Transfrontal and Transsphenoidal Approaches to Pediatric Craniopharyngioma: A National Perspective. Pediatr Neurosurg. 2017 Feb 23. [QxMD MEDLINE Link].
van Iersel L, Meijneke RWH, Schouten-van Meeteren AYN, et al. The development of hypothalamic obesity in craniopharyngioma patients: A risk factor analysis in a well-defined cohort. Pediatr Blood Cancer. 2018 May. 65 (5):e26911. [QxMD MEDLINE Link].
Abdu TA, Elhadd TA, Neary R, Clayton RN. Comparison of the low dose short synacthen test (1 microg), the conventional dose short synacthen test (250 microg), and the insulin tolerance test for assessment of the hypothalamo-pituitary-adrenal axis in patients with pituitary disease. J Clin Endocrinol Metab. 1999 Mar. 84(3):838-43. [QxMD MEDLINE Link].
Streeten DH. Shortcomings in the low-dose (1 microg) ACTH test for the diagnosis of ACTH deficiency states. J Clin Endocrinol Metab. 1999 Mar. 84(3):835-7. [QxMD MEDLINE Link].
Chanoine JP, Rebuffat E, Kahn A, et al. Glucose, growth hormone, cortisol, and insulin responses to glucagon injection in normal infants, aged 0.5-12 months. J Clin Endocrinol Metab. 1995 Oct. 80(10):3032-5. [QxMD MEDLINE Link].
Fischli S, Jenni S, Allemann S, et al. Dehydroepiandrosterone sulfate in the assessment of the hypothalamic-pituitary-adrenal axis. J Clin Endocrinol Metab. 2008 Feb. 93(2):539-42. [QxMD MEDLINE Link].
Coutant R, Biette-Demeneix E, Bouvattier C, et al. Baseline inhibin B and anti-Mullerian hormone measurements for diagnosis of hypogonadotropic hypogonadism (HH) in boys with delayed puberty. J Clin Endocrinol Metab. 2010 Dec. 95(12):5225-32. [QxMD MEDLINE Link].
Carel JC, Tresca JP, Letrait M, et al. Growth hormone testing for the diagnosis of growth hormone deficiency in childhood: a population register-based study. J Clin Endocrinol Metab. 1997 Jul. 82(7):2117-21. [QxMD MEDLINE Link].
Marin G, Domene HM, Barnes KM, et al. The effects of estrogen priming and puberty on the growth hormone response to standardized treadmill exercise and arginine-insulin in normal girls and boys. J Clin Endocrinol Metab. 1994 Aug. 79(2):537-41. [QxMD MEDLINE Link].
Li G, Shao P, Sun X, Wang Q, Zhang L. Magnetic resonance imaging and pituitary function in children with panhypopituitarism. Horm Res Paediatr. 2010. 73(3):205-9. [QxMD MEDLINE Link].
DeVile CJ, Stanhope R. Hydrocortisone replacement therapy in children and adolescents with hypopituitarism. Clin Endocrinol (Oxf). 1997 Jul. 47(1):37-41. [QxMD MEDLINE Link].
Giagulli VA, Castellana M, Perrone R, Guastamacchia E, Iacoviello M, Triggiani V. GH Supplementation Effects on Cardiovascular Risk in GH Deficient Adult Patients: A Systematic Review and Meta-analysis. Endocr Metab Immune Disord Drug Targets. 2017 Nov 16. 17 (4):285-96. [QxMD MEDLINE Link].
Feferkorn I, Badeghiesh A, Baghlaf H, Dahan MH. Pregnancy outcomes in women with panhypopituitarism: a population-based study. Reprod Biomed Online. 2022 Mar. 44 (3):532-7. [QxMD MEDLINE Link].
Schonberger J, Eckenweiler M, Klotz KA, et al. Facilitation of drug-resistant epilepsy and catastrophic status epilepticus in children with combined pituitary hormone deficiency. Eur J Paediatr Neurol. 2021 Jul. 33:99-105. [QxMD MEDLINE Link].
Bates AS, Van't Hoff W, Jones PJ, Clayton RN. The effect of hypopituitarism on life expectancy. J Clin Endocrinol Metab. 1996 Mar. 81(3):1169-72. [QxMD MEDLINE Link].
Rosen T, Bengtsson BA. Premature mortality due to cardiovascular disease in hypopituitarism. Lancet. 1990 Aug 4. 336(8710):285-8. [QxMD MEDLINE Link].
Twickler TB, Wilmink HW, Schreuder PC, et al. Growth hormone (GH) treatment decreases postprandial remnant-like particle cholesterol concentration and improves endothelial function in adult-onset GH deficiency. J Clin Endocrinol Metab. 2000 Dec. 85(12):4683-9. [QxMD MEDLINE Link].
Claessen KM, Appelman N, Pereira AM, Joustra SD, Mutsert R, Gast KB, et al. Abnormal metabolic phenotype in middle-aged Growth Hormone Deficient (GHD) adults despite long-term recombinant human GH (rhGH) replacement. Eur J Endocrinol. 2013 Nov 11. [QxMD MEDLINE Link].
Hoffman RP. Growth hormone (GH) treatment does not restore endothelial function in children with GH deficiency. J Pediatr Endocrinol Metab. 2008 Apr. 21(4):323-8. [QxMD MEDLINE Link].
Lanes R, Soros A, Flores K, Gunczler P, Carrillo E, Bandel J. Endothelial function, carotid artery intima-media thickness, epicardial adipose tissue, and left ventricular mass and function in growth hormone-deficient adolescents: apparent effects of growth hormone treatment on these parameters. J Clin Endocrinol Metab. 2005 Jul. 90(7):3978-82. [QxMD MEDLINE Link].
O'Neal D, Hew FL, Sikaris K, Ward G, Alford F, Best JD. Low density lipoprotein particle size in hypopituitary adults receiving conventional hormone replacement therapy. J Clin Endocrinol Metab. 1996 Jul. 81(7):2448-54. [QxMD MEDLINE Link].
Santoro SG, Guida AH, Furioso AE, Glikman P, Rogozinski AS. Panhypopituitarism due to Wegener's granulomatosis. Arq Bras Endocrinol Metabol. 2011 Oct. 55(7):481-5. [QxMD MEDLINE Link].
Gazzaruso C, Gola M, Karamouzis I, Giubbini R, Giustina A. Cardiovascular Risk in Adult Patients With Growth Hormone (GH) Deficiency and Following Substitution with GH--An Update. J Clin Endocrinol Metab. 2013 Nov 11. [QxMD MEDLINE Link].
Carel JC, Ecosse E, Landier F, Meguellati-Hakkas D, Kaguelidou F, Rey G, et al. Long-term mortality after recombinant growth hormone treatment for isolated growth hormone deficiency or childhood short stature: preliminary report of the French SAGhE study. J Clin Endocrinol Metab. 2012 Feb. 97(2):416-25. [QxMD MEDLINE Link].
Sävendahl L, Maes M, Albertsson-Wikland K, Borgström B, Carel JC, Henrard S, et al. Long-term mortality and causes of death in isolated GHD, ISS, and SGA patients treated with recombinant growth hormone during childhood in Belgium, The Netherlands, and Sweden: preliminary report of 3 countries participating in the EU SAGhE study. J Clin Endocrinol Metab. 2012 Feb. 97(2):E213-7. [QxMD MEDLINE Link].
Mo D, Hardin DS, Erfurth EM, Melmed S. Adult mortality or morbidity is not increased in childhood-onset growth hormone deficient patients who received pediatric GH treatment: an analysis of the Hypopituitary Control and Complications Study (HypoCCS). Pituitary. 2013 Oct 12. [QxMD MEDLINE Link].
Charmandari E, Lichtarowicz-Krynska EJ, Hindmarsh PC, et al. Congenital adrenal hyperplasia: management during critical illness. Arch Dis Child. 2001 Jul. 85(1):26-8. [QxMD MEDLINE Link].
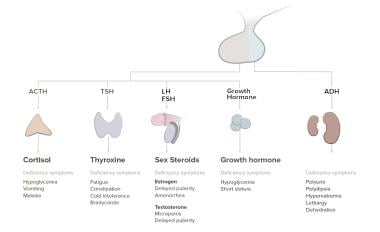 Pathophysiology of panhypopituitarism.
Pathophysiology of panhypopituitarism.








