Background
Ventral abdominal body wall defects comprise a group of congenital malformations that includes gastroschisis and omphalocele, which are relatively common, and ectopia cordis, bladder exstrophy, and cloacal exstrophy, which are extremely rare. Gastroschisis is the congenital anomaly most frequently encountered by pediatric surgeons, and the incidence is rising. The incidence of omphaloceles, however, has remained relatively constant. [1]
The reported incidence of abdominal wall defects in the United States is estimated to be the following:
-
Gastroschisis: 1 case in 2,229 births (about 1,871 infants each year) [2]
-
Omphalocele: 1 case in 5,386 births (about 775 babies annually) [2]
-
Bladder exstrophy: 1 case in 50,000 births [3]
-
Ectopia cordis: 1 case in 125,000 births [4]
-
Cloacal exstrophy: 1 case in 10,000 - 70,000 to 1 case in 200,000 - 400,000 births [5]
There is also anatomic variation regarding the size and location of the abdominal wall defect, as well as a spectrum of associated anomalies:
-
Gastroschisis: Small paraumbilical defect, associated intestinal abnormalities
-
Omphalocele: Central umbilical defect of varying size; omphaloceles may be syndromic (genetic) or have associated system abnormalities
-
Epigastric omphalocele (pentalogy): Associated diaphragmatic, sternal, pericardial, and heart abnormalities
-
Bladder exstrophy: Lower abdominal wall and pelvis, associated genitourinary abnormalities
-
Cloacal exstrophy: Bladder exstrophy plus an imperforate anus
Furthermore, within each category, there is a spectrum of severity with regard to the initial problem and the anticipated secondary problems.
An infant with gastroschisis may have intestinal dysfunction. Prolonged exposure to amniotic fluid may cause mucosal or muscularis injury, although the etiology of bowel injury in gastroschisis is unclear. If noxious elements in the amniotic fluid were responsible, amnioexchange should be therapeutic. Unfortunately, the anticipated benefit has not occurred. [6] Or, potentially or a small abdominal wall defect may constrict the mesentery, causing ischemic injury or actual necrosis.
Intestinal obstruction, from inflammatory adhesions or from Ladd bands, a component of malrotation, may complicate the infant's recovery. Malrotation occurs in the setting of developmental anomalies, in which the intestine fails to return to the nascent abdominal cavity, such as congenital diaphragmatic hernia as well as abdominal wall defects. Midgut volvulus, the most feared complication of malrotation, is theoretically possible but unlikely because of postsurgical adhesions. The appendix is abnormally located in the upper abdomen in patients with malrotation; however, the availability of computed tomography (CT) scanning to evaluate the abdomen makes this less of an issue. Children with gastroschisis may suffer from gastroesophageal reflux; the prevalence of Hirschsprung disease is also increased.
See the Medscape Drugs & Diseases articles Omphalocele Imaging and Gastroschisis Imaging, as well as information from the Children's Hospital of Philadelphia on Omphalocele and Gastroschisis.
See also the Critical Images slideshow 11 Abdominal Emergencies in Infants.
Pathophysiology
Embryology
Following fertilization, cellular division generates a hollow sphere, bisected by a bilaminar plate composed of epiblast and hypoblast, which abut the amnion and yolk sac cavities respectively. [7] The epiblast cells form the embryo; the hypoblast develops into the placenta. The bilaminar disc is divided axially by the primitive streak, at the apex of which is the primitive node. Epiblast cells pour into the primitive node, converting the bilaminar disc into a disk with three germ cell layers. Invagination occurs along the primitive streak, giving the embryo (in cross-section) the appearance of an omega. The curved portion of the omega fuses, forming the neural tube (central nervous system [CNS]) and displacing the notochord ventrally. The pinched-off tissue, dorsal to the neural tube, becomes epiderm. Laterally, the mesoderm undergoes differentiation: para-axial (peripheral nervous system), intermediate (gonads and kidneys), and lateral, which further divides into splanchnic (gastrointestinal [GI] tract) and somatic (body wall). The splanchnic mesoderm and endoderm fuse ventrally to form a tube (the GI tract); the somatic mesoderm and epiderm layers fuse anteriorly forming the anterior body wall and abdominal cavity (intraembryonic coelom). The embryo is surrounded by amnion, which encloses the extra-embryonic coelom. [8]
During the sixth week of development, rapid growth of the liver and intestines causes herniation of the midgut into the amniotic cavity. By the 10th week, the abdominal cavity is sufficiently large to accommodate return of the midgut. Rotation and fixation of the proximal and distal midgut (duodenum and ascending colon) occurs upon their return.
As the hindgut elongates, ingrowth of mesoderm forms the urorectal septum. Mesoderm also invades the cloacal membrane, uniting the genital tubercles and creating the urogenital sinus.
The body folds unite where the amnion invests the yolk sac and body stalk. Fusion is a complex process: A surface glycoprotein promotes adhesion between opposing segments of the nascent body wall; migration and apoptosis (planned cellular death) knit together the cephalic, caudal, and lateral folds about the attenuated yolk sac, causing constriction of the umbilical ring.
Omphaloceles and gastroschisis
Omphaloceles
In infants with omphaloceles, the intestines do not return to the abdominal cavity; rather, they remain within the extra-embryonic coelom (amniotic cavity) bounded by the umbilical ring. [9] (See the image below.)
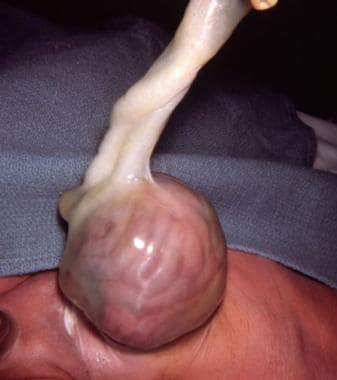 Pediatric omphalocele and gastroschisis (abdominal wall defects). An intact omphalocele in an infant is shown.
Pediatric omphalocele and gastroschisis (abdominal wall defects). An intact omphalocele in an infant is shown.
There is evidence to suggest that omphaloceles have a genetic etiology, as follows:
-
Omphaloceles are associated with increased maternal age.
-
Omphaloceles occur in twins, consecutive children, and different generations of the same family.
-
Omphaloceles are associated with trisomy 13, 18, and 21 (in 25%-50 % of cases) and with Beckwith-Wiedemann syndrome
Gastroschisis
In gastroschisis, there appears to be a weakness in the body wall (caused by defective ingrowth of mesoderm, or impaired midline fusion, or inappropriate apoptosis) that allows the intestines to herniate through this defect into the amniotic cavity. (See the following image.)
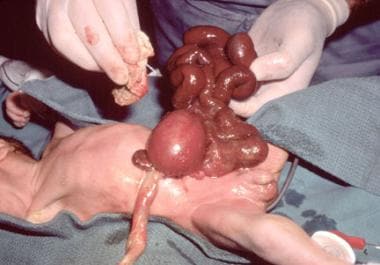 Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative photograph reveals and infant with gastroschisis.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative photograph reveals and infant with gastroschisis.
Gastroschisis occurs in young mothers with low gravida; it is associated with prematurity and small-for-gestational-age (SGA) infants, and denotes in utero growth retardation.
The clustering of cases (number and severity) suggests a multifactorial etiology, including environmental factors acting upon susceptible hosts.
Other abdominal wall defects
Issues to consider in understanding the spectrum of body wall defects include the following:
-
Is a sac is present?
-
Is it ruptured?
-
Are there associated anomalies? (This suggests omphalocele, as they are twice as more prevalent in omphalocele than gastroschisis. [10] )
Associated anomalies include:
-
Congenital heart disease
-
Cleft palate
-
Musculoskeletal abnormalities
-
Dental malocclusion
-
Intestinal atresia
-
Patent omphalomesenteric duct remnant (looks like a stoma at the base of the umbilical cord)
Hernias of the umbilical cord
In hernias of the umbilical cord, the umbilical ring is oversized, but the relationships of amnion to yolk sac and connecting stalk are normal. (See the image below.)
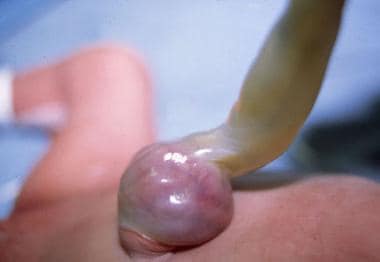 Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph depicts an umbilical cord hernia in an infant.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph depicts an umbilical cord hernia in an infant.
Urachal remnants and omphalomesenteric duct malformations
Urachal remnants [11] and omphalomesenteric duct malformations [12] are the result of insufficient apoptotic cell death in the urachus and yolk stalk. (See the following images.)
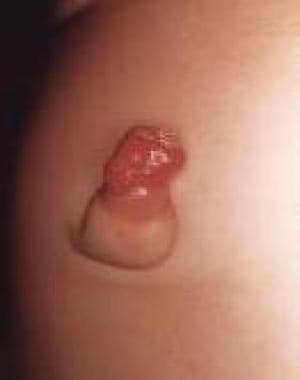 Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows an omphalomesenteric duct remnant presenting as an "umbilical granuloma."
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows an omphalomesenteric duct remnant presenting as an "umbilical granuloma."
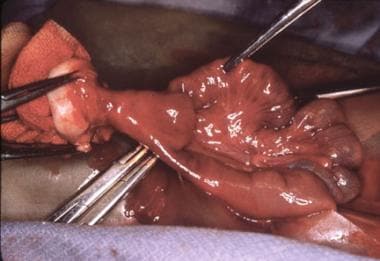 Pediatric omphalocele and gastroschisis (abdominal wall defects). An intraoperative finding is a patent omphalomesenteric duct, which is being excised.
Pediatric omphalocele and gastroschisis (abdominal wall defects). An intraoperative finding is a patent omphalomesenteric duct, which is being excised.
Bladder exstrophy
The bladder develops between the fifth and ninth gestational weeks (postfertilization). [13] By 10 weeks, urine is produced and mixes with the amniotic fluid; this is crucial for normal lung development.
In healthy infants, the bladder is visible on ultrasonography toward the end of the first trimester. In these patients, the bladder is open with exposed mucosa, forming a protruding disc below the umbilical cord, which is displaced caudally. The pelvic rami are spread apart; consequently, the pelvis is shallow and lacks depth. Hence, the developing bladder, urethra, vagina, and rectum are displaced anteriorly. The abnormal position of these organs interferes with normal development of the lower abdominal wall. (See the images below.)
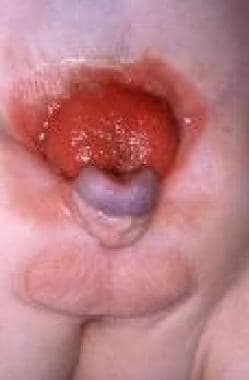 Pediatric omphalocele and gastroschisis (abdominal wall defects). Bladder exstrophy and epispadias is shown in an infant. Note the appearance of the bladder mucosa, indicating chronic inflammation.
Pediatric omphalocele and gastroschisis (abdominal wall defects). Bladder exstrophy and epispadias is shown in an infant. Note the appearance of the bladder mucosa, indicating chronic inflammation.
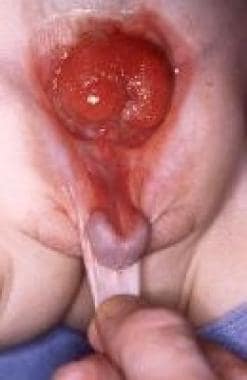 Pediatric omphalocele and gastroschisis (abdominal wall defects). This is another view demonstrating the epispadias shown in the previous image.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This is another view demonstrating the epispadias shown in the previous image.
Prune-belly syndrome
Prune-belly syndrome includes dilatation of the ureter, collecting system, and bladder; undescended testes; and other abnormalities. This syndrome is caused by increased "apoptotic" cell death in the body-wall placode or inadequate migration of mesodermal cells, with retention of yolk sac elements. [13]
Characteristics of prune-belly syndrome include the following:
-
There is attenuation of the abdominal musculature.
-
Muscle fibers are absent and are replaced by thick collagenous aponeuroses.
-
Paradoxically, hypoplasia of the abdominal wall contrasts with hypertrophy of the bladder wall, which may cause bladder neck obstruction and the characteristic megaureters and dilated renal collecting system.
-
Faulty intercellular electrical conduction causes disordered muscular contraction and ineffective ureteric peristalsis.
-
Approximately 95% of infants with prune-belly syndrome are male; the absence of prostatic and seminal fluid precludes normal sperm development and causes infertility, as may the associated cryptorchidism.
Cloacal exstrophy
The urorectal septum divides the cloaca into the urogenital sinus and the rectum. Defective enfolding of the embryo's caudal pole, and deficient incorporation of the yolk sac and allantois into the urogenital sinus, leads to malformation of the external genitalia.
The cloaca persists in the absence of mesodermal ingrowth. Differentiation of the genitourinary system and hindgut are arrested, and development of the lower abdominal wall is thwarted.
Cloacal exstrophy is associated with mutations in the homeobox genes. (See the following images.)
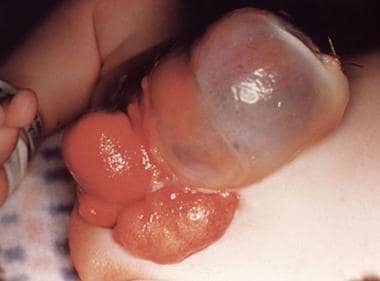 Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph shows cloacal exstrophy in an infant.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph shows cloacal exstrophy in an infant.
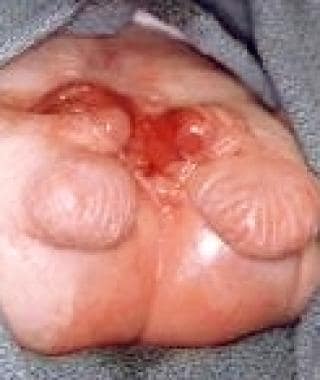 Pediatric omphalocele and gastroschisis (abdominal wall defects). Note the bifid genitalia in this infant with cloacal exstrophy.
Pediatric omphalocele and gastroschisis (abdominal wall defects). Note the bifid genitalia in this infant with cloacal exstrophy.
Etiology
In rat studies, folic acid deficiency, hypoxia, and salicylates cause abdominal wall defects to develop, but the clinical significance of these experiments is conjectural.
Elevation of maternal serum alpha-fetoprotein (MSAFP) is associated with omphalocele and gastroschisis. An elevated MSAFP warrants ultrasonographic evaluation to determine if structural abnormalities are present in the fetus. If the study is suspicious for an omphalocele, amniocentesis is indicated to determine the presence of an associated genetic abnormality.
Polyhydramnios occurs in association with intestinal atresia, which may complicate gastroschisis. If polyhydramnios is identified by fetal ultrasonography, the mother should be referred to a tertiary care facility for optimal care of her newborn. Generally, newborns requiring surgery are best managed at tertiary care centers.
Epidemiology
United States data
Analysis of 2012-2016 data from 30 US population-based birth defect surveillance programs by the National Birth Defects Prevention Network (NBDPN) focused on abdominal wall defects, specifically gastroschisis and omphalocele. It found an overall prevalence of 4.3 per 10,000 live births for gastroschisis and 2.1 per 10,000 live births for omphalocele. [10] Gastroschisis was more frequent among mothers younger than 25 years and more likely to occur when mothers had low or normal prepregnancy weights; it was half as likely as omphalocele to occur in conjunction with other birth defects. Omphalocele was more common in mothers older than 40 years, more likely to occur in overweight/obese mothers, and twice as likely as gastroschisis to concomitantly occur with other birth defects. [10]
Allman et al evaluated retrospective discharge data (1997-2015) for the prevalence of infants with gastroschisis in US neonatal intensive care units (NICUs). Of 1,158,755 total discharges, 6,023 infants (5.2 per 1000 discharges) had gastroschisis and 1,885 (1.6 per 1000 discharges) had an omphalocele. [14] The rate of gastroschisis increased from 2.9 to 6.4 per 1000 discharges over a 12-year period (1997-2008), gradually declined over the next 4 years (2008-2011) from 6.4 to 4.7 per 1000 discharges, and then remained stable thereafter. The rate of omphalocele was stable over the same time periods, at 1-2 per 1000 discharges. [14]
International data
The combined incidence of omphalocele and gastroschisis is 1 case per 3,500 births. Epidemiologic data compiled over the last four to five decades show that the incidence of omphalocele has remained constant, whereas that of gastroschisis has increased three- to four-fold. See the table below.
Table 1. Incidence Rates for Gastroschisis and/or Omphaloceles in Various Regions and Time Periods. [1, 15, 16, 17] (Open Table in a new window)
Country |
Time Period / Incidence |
Time Period / Incidence |
Japan |
1975-1980 |
1996-1997 |
Gastroschisis |
1/77,000 |
1/20,000 |
Omphalocele |
1/30,000 |
1/27,000 |
|
|
|
England and Wales |
1987 |
1991 |
Gastroschisis |
1/10,000 |
2/10,000 |
Omphalocele |
1/10,000 |
1/12,500 |
|
|
|
Galveston, Texas |
1983 |
2002 |
Gastroschisis |
1/4000 |
1/900 |
|
|
|
England and Wales |
1995 |
2005 |
Gastroschisis |
1/7500 |
1/2500 |
Race- and sex-related demographics
Neither gastroschisis nor omphalocele has a geographic or racial predilection. The Texas data indicate that gastroschisis occurs in diminishing frequency among Latinos, white persons, and black persons. [1]
The male-to-female ratio is about 1.5:1.
Prognosis
Omphalocele
Care of infants with omphaloceles may be simple or complex, depending upon the size of the defect, and the presence and severity of associated problems.
Infants with giant omphaloceles, in which the liver is centrally located and contained within the omphalocele sac, usually have small, bell-shaped thoraces and pulmonary hypoplasia. These babies frequently require tracheostomies and assisted ventilation. Even so, patience and optimism should be encouraged, as growth of the thoracic cavity and maturation of the lung may eventuate in a gratifying outcome.
Gastroschisis
The prognosis of infants with gastroschisis depends upon the severity of associated problems, such as prematurity, intestinal atresia and dysfunction, and possibly short gut. A population-based cohort study from 28 pediatric surgical centers in the United Kingdom and Ireland analyzed the 1-year outcomes of infants with gastroschisis and found that infants with complex gastroschisis required longer hospital stays and had more complications than infants with simple gastroschisis. [18] Classifying infants with gastroschisis into "simple" versus "complex" (macroscopic intestinal abnormalities) may be a reliable predictor of outcome. [18] A separate population-based study of 502 Australian infants with abdominal wall defects (166 omphalocele, 336 gastroschisis) reported similar findings of longer hospital stays and parenteral nutrition as well as higher rates of infection but lower overall mortality in infants with gastroschisis compared to those with omphalocele. [19]
The prognosis has been greatly improved by antenatal sonography, which allows identification of infants with abdominal wall defects and referral to tertiary centers where prenatal, obstetrical, and pediatric surgical expertise is available. [15, 16, 20]
Morbidity/mortality
The mortality of omphaloceles relative to gastroschisis is 8:1. Irreversible pulmonary hypertension and right heart failure is the usual terminal event.
In a 2018 literature review of 23 articles comprising 396 giant omphaloceles, the outcome was lethal in in nearly 23% (n = 90) of neonates, with sepsis the primary cause in more than half of these patients (56.6%; n = 51). [21] Two predictors of mortality were pulmonary hypoplasia and respiratory failure; prematurity and ruptured sacs were also implicated. [21]
Factors adversely influencing the management of infants with gastroschisis are as follows:
-
Prematurity and low birth weight
-
Hypothermia (exposure of the intestine to the ambient environment)
-
Dehydration (gastrointestinal losses, in addition to the above factors)
-
Sepsis (open wound)
-
Hypoglycemia (stress with little metabolic reserve)
-
In utero growth restriction (protein loss from the extruded intestines)
-
Oligohydramnios
-
Fetal distress and birth asphyxia
-
Injury to the intestines during delivery (tearing or cutting the bowel or mesentery) or transport (allowing the eviscerated intestines to fall alongside the infant, stretching or torquing the mesenteric vessels) [22]
Improvements in respiratory care, pharmacology (antibiotics and total parenteral nutrition), anesthesia, transport, and surgery have increased the survival rates for these infants from 60% during the 1960s to more than 90% in more recent years. [15, 16, 20]
See the images below.
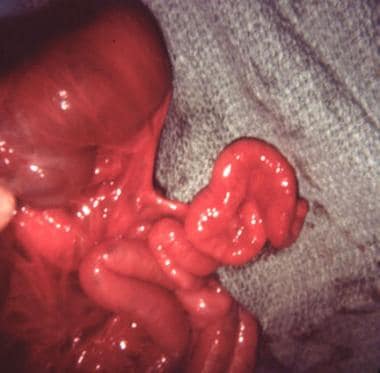 Pediatric omphalocele and gastroschisis (abdominal wall defects). Gastroschisis and associated intestinal atresia is shown in an infant.
Pediatric omphalocele and gastroschisis (abdominal wall defects). Gastroschisis and associated intestinal atresia is shown in an infant.
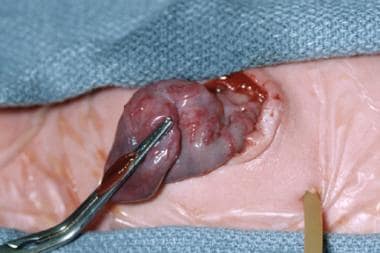 Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative image reveals gastroschisis and colon atresia in an infant. The bulbous proximal end of the atretic colon is excised, and a colostomy is created at the abdominal wall defect. An anastomosis of the proximal, dilated colon to the distal microcolon (in view of its small caliber) would not function properly. The colostomy can be closed 4-6 weeks later.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative image reveals gastroschisis and colon atresia in an infant. The bulbous proximal end of the atretic colon is excised, and a colostomy is created at the abdominal wall defect. An anastomosis of the proximal, dilated colon to the distal microcolon (in view of its small caliber) would not function properly. The colostomy can be closed 4-6 weeks later.
Long-term morbidity from gastroschisis is related to intestinal dysmotility (pseudointestinal obstruction), malabsorption (mucosal injury), short gut, and gastroesophageal reflux disease. Difficulty obtaining wound closure contributes to morbidity by prolonging intestinal dysfunction (ileus) and creating ventral hernias, which may require surgical repair. [23, 24, 6, 25]
The following scenarios may cause short-gut syndrome, in which the intestinal length is inadequate:
-
An antenatal mesenteric vascular accident may cause intestinal atresia.
-
Constriction of the extruded intestine's mesentery by a small abdominal wall defect may cause in utero gut infarction ("closing gastroschisis").
-
Tethering the eviscerated bowel's mesentery impairs blood flow and may cause vascular injury. [22]
-
Excessive tension from closure of the abdominal wall defect results in the "abdominal compartment syndrome," in which the intra-abdominal pressure exceeds the splanchnic perfusion pressure and nutritive blood flow ceases.
Closed-loop obstructions, in which both efferent and afferent limbs of the intestine are blocked, occur in volvulus (twisting of the entire midgut and its mesentery), or a single loop of intestine may flip around a point of fixation, such as an adhesion to the abdominal wall. This causes unrelieved distention and ischemic injury of the intestines (ie, "strangulation obstruction"). [26]
The injury produced by antenatal exposure of the intestine to amniotic fluid (mucosal and muscular) leads to diminished absorption and impaired peristalsis, which compounds the crippling effect of the diminished length of short-gut syndrome.
The care of infants with short-gut syndrome has improved with innovations in parenteral and enteral nutrition, venous access devices, prevention and early treatment of catheter sepsis, innovative surgical procedures to optimize gut length, and aggressive treatment of bacterial overgrowth in stagnant loops of intestine. Infants with short-gut syndrome from gastroschisis account for a substantial number of children who undergo intestinal transplantation. [27, 28]
See the images below.
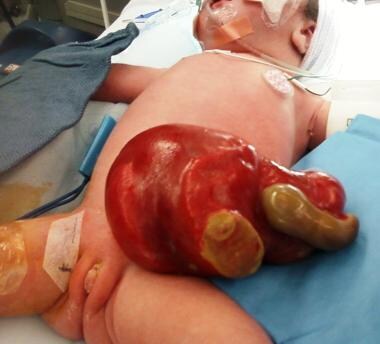 Pediatric omphalocele and gastroschisis (abdominal wall defects). In this infant, gastroschisis has been complicated by jejunal atresia and loss of the entire distal small bowel (the grey tissue).
Pediatric omphalocele and gastroschisis (abdominal wall defects). In this infant, gastroschisis has been complicated by jejunal atresia and loss of the entire distal small bowel (the grey tissue).
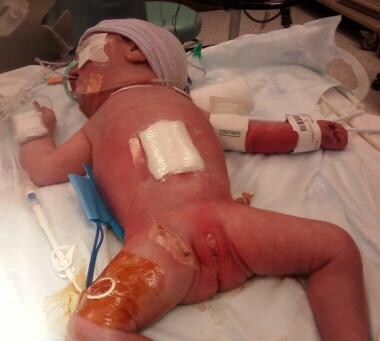 Pediatric omphalocele and gastroschisis (abdominal wall defects). Following lysis of adhesions and tubularization of the viable, mesenteric portion of the proximal jejunum, the eviscerated viscera are reduced and the gastroschisis abdominal wall defect is closed.
Pediatric omphalocele and gastroschisis (abdominal wall defects). Following lysis of adhesions and tubularization of the viable, mesenteric portion of the proximal jejunum, the eviscerated viscera are reduced and the gastroschisis abdominal wall defect is closed.
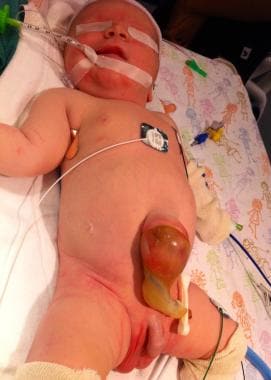 Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant had a small omphalocele sac whose contents were liver and gall bladder.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant had a small omphalocele sac whose contents were liver and gall bladder.
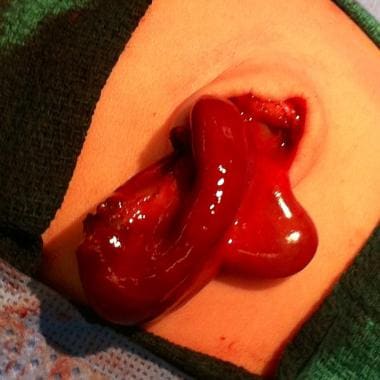 Pediatric omphalocele and gastroschisis (abdominal wall defects). The omphalocele sac contains liver and gall bladder.
Pediatric omphalocele and gastroschisis (abdominal wall defects). The omphalocele sac contains liver and gall bladder.
 Pediatric omphalocele and gastroschisis (abdominal wall defects). A completed repair is shown, simulating an umbilicus.
Pediatric omphalocele and gastroschisis (abdominal wall defects). A completed repair is shown, simulating an umbilicus.
Infants with giant omphaloceles usually have small, bell-shaped thoraces and minimal pulmonary reserve. Repair of the omphalocele may precipitate respiratory failure by limiting diaphragmatic excursion.
Occasionally, infants are encountered with anomalies whose adverse effects are additive, such as a giant omphalocele plus a diaphragmatic hernia. Pulmonary hypoplasia occurs in both conditions; when they occur together, their effects are so severe as to preclude survival, despite extracorporeal membrane oxygenation (ECMO) support.
Even with successful repair of a giant omphalocele, the liver remains located in the midepigastrium, where it lacks the normal protection afforded by the lower rib cage and where it is more vulnerable to injury. See the following image.
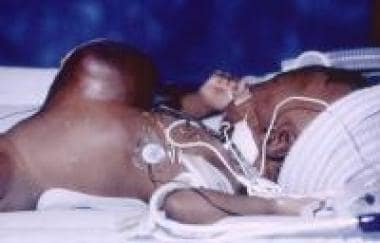 Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows a giant omphalocele in an infant, in which the liver assumes an ectopic position in the epigastrium.
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows a giant omphalocele in an infant, in which the liver assumes an ectopic position in the epigastrium.
A study by Corey et al indicated that compared with infants with gastroschisis, those with omphalocele have a higher incidence of other anomalies, are more likely to have pulmonary hypertension, and have a higher mortality rate. [29] In the study, which involved 4,687 infants with gastroschisis and 1,448 with omphalocele, the investigators found that 35% of the patients with omphalocele had at least one other anomaly, as compared with 8% of those with gastroschisis. The odds ratios for pulmonary hypertension and mortality in infants with omphalocele compared with those with gastroschisis were 7.78 and 6.81, respectively. [29]
Complications
Babies whose omphaloceles are treated conservatively ("paint and wait") have increased caloric requirements, because of their large open wound. Positive nitrogen balance is restored following skin closure.
Prolonged parenteral nutrition can cause hepatotoxicity, manifested by cholestasis and hepatomegaly, which may complicate a staged closure of a giant omphalocele. Omega-3 fatty acids (Omegaven) reportedly may reverse "intestinal failure associated liver disease."
Infants with giant omphaloceles have pulmonary hypoplasia in addition to diminutive thoraces; they may require tracheotomies and ventilatory support. Abdominal wall closure transiently increases intra-abdominal pressure; paradoxically, reconstituting the infant's torso improves muscular function and may enable ventilator weaning.
Infants with giant omphaloceles have an increased risk of sepsis, [21] because of the open wound and need for ventilatory support and central venous access.
Patient Education
Instruct parents of infants with gastroschisis or omphalocele regarding the significance (ominous) of bilious emesis, because this may indicate that adhesive small-bowel obstruction or midgut volvulus has occurred.
Also inform parents that their child's appendix is located in an unusual location and that computed tomography scanning is the most reliable way to diagnose acute appendicitis.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). An intact omphalocele in an infant is shown.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph depicts an umbilical cord hernia in an infant.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative photograph reveals and infant with gastroschisis.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative image reveals a ruptured omphalocele in an infant.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Gastroschisis and associated intestinal atresia is shown in an infant.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This intraoperative image reveals gastroschisis and colon atresia in an infant. The bulbous proximal end of the atretic colon is excised, and a colostomy is created at the abdominal wall defect. An anastomosis of the proximal, dilated colon to the distal microcolon (in view of its small caliber) would not function properly. The colostomy can be closed 4-6 weeks later.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Note the enlarged tongue in this infant with Beckwith-Wiedemann syndrome.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant has pentalogy of Cantrell: an epigastric omphalocele, cleft sternum, anterior (retrosternal) diaphragmatic hernia of Morgagni, absent pericardium, and cardiac defects (ectopia cordis, ventricular septal defects).
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image demonstrates silo closure in an infant with gastroschisis.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Silon sheets are pulled over the omphalocele sac, elevating the rectus muscles, and, because of their attachment to the costal arch, expanding the thoracic cavity. The Silon sheets are removed and replaced by a permanent Gore-Tex patch that is covered by skin flaps.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant has a giant omphalocele that was treated with topical agents for several weeks. The omphalocele sac will absorb, leaving granulation tissue that gradually epithelializes.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). The omphalocele sac was adherent to the protuberant liver in an infant. It was covered with Gore-Tex so that gradual reduction could be effected.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). The Gore-Tex sheet is imbricated, gradually reducing the liver into the abdominal cavity: The rectus muscles are pulled over the liver. The skin and subcutaneous tissues become adherent to the Gortex; the next step is to freshen the wound margins (excise the skin edge and the exposed Gortex) and suture the composite layers together.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Final skin closure of the giant omphalocele was delayed because the baby developed respiratory distress. Unfortunately, the patch became infected and was removed. Later, bipedicled flank flaps were used to close the giant omphalocele, but reduction was lost.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Split-thickness skin grafts were applied to the flank wounds resulting from mobilization of the bipedicle flaps.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant has prune-belly syndrome.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Note the laxity of the abdominal wall in this infant with prune-belly syndrome.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph shows cloacal exstrophy in an infant.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Note the bifid genitalia in this infant with cloacal exstrophy.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). In the repair of cloacal exstrophy, the cecal plate in the middle of the bifid bladder is excised and used to create an ostomy, and the bladder halves are approximated.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Closure of the bladder exstrophy is shown.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Bladder exstrophy and epispadias is shown in an infant. Note the appearance of the bladder mucosa, indicating chronic inflammation.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This is another view demonstrating the epispadias shown in the previous image.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image reveals isolated epispadias in an infant.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). An intraoperative finding is a patent omphalomesenteric duct, which is being excised.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Closure of a giant omphalocele with an Alloderm patch is shown.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image was obtained 2 months after implantation, revealing epithelialization of the Alloderm patch.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Eight months after implantation, epithelialization is nearly complete, but a huge ventral hernia has developed.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows a giant omphalocele in an infant, in which the liver assumes an ectopic position in the epigastrium.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). In this infant, gastroschisis has been complicated by jejunal atresia and loss of the entire distal small bowel (the grey tissue).
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Following lysis of adhesions and tubularization of the viable, mesenteric portion of the proximal jejunum, the eviscerated viscera are reduced and the gastroschisis abdominal wall defect is closed.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). The radiograph shows the intestine following multiple serial transverse enteroplasty (STEP) procedures.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant has gastroschisis and colon atresia, with the proximal end open. An ostomy is brought out of the silo.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Inflammatory distortion of the extruded intestine is shown. There appears to be an associated atresia (the dilated intestine), but the dilatation resolved in concert with resolution of the inflammation.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). A silo is fashioned from Silon sheets, in case reduction of the extruded intestine cannot be achieved without unduly elevating the intra-abdominal pressure.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). The sac is removed, and the abdominal wall defect is closed around the in utero colostomy.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This infant had a small omphalocele sac whose contents were liver and gall bladder.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). The omphalocele sac contains liver and gall bladder.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). A completed repair is shown, simulating an umbilicus.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This child, approximately age 30 months in the photo, had a successful repair. Her clinical course is described above.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows the appearance of the dilated bowel prior to performing the serial transverse enteroplasty (STEP) procedure.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This is the intestine as it appeared after the serial transverse enteroplasty (STEP) procedure.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This is the intestine as it appeared after the serial transverse enteroplasty (STEP) procedure.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This image shows an omphalomesenteric duct remnant presenting as an "umbilical granuloma."
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Rather than pushing the extruded viscera into the diminutive abdominal cavity, traction is applied to the abdominal wall and skin flaps. This stimulates growth and facilitates reduction of the extruded visceral and ultimate closure of the abdominal wall defect. Courtesy of Russell Jennings, MD, Boston Children's Hospital.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). This photograph depicts progress in reducing the extruded viscera utilizing Dr Jennings's innovative technique. Image courtesy of Russell Jennings, MD, Boston Children's Hospital.
-
Pediatric omphalocele and gastroschisis (abdominal wall defects). Reduction of the extruded viscera is gradually achieved. Courtesy of Russell Jennings, MD, Boston Children's Hospital.
Tables
Country |
Time Period / Incidence |
Time Period / Incidence |
Japan |
1975-1980 |
1996-1997 |
Gastroschisis |
1/77,000 |
1/20,000 |
Omphalocele |
1/30,000 |
1/27,000 |
|
|
|
England and Wales |
1987 |
1991 |
Gastroschisis |
1/10,000 |
2/10,000 |
Omphalocele |
1/10,000 |
1/12,500 |
|
|
|
Galveston, Texas |
1983 |
2002 |
Gastroschisis |
1/4000 |
1/900 |
|
|
|
England and Wales |
1995 |
2005 |
Gastroschisis |
1/7500 |
1/2500 |







